



ISSUE TOPIC: TRENDS IN QUALITY CONTROL AND STANDARDISATION OF BIOLOGICALS

INTRODUCTION. Currently, there are no harmonised regulatory requirements for the quality control of human somatic cell therapy and tissue-engineered medicinal products that contain differentiated cells derived from induced pluripotent stem cells (iPSCs). This lack of uniform requirements underscores the need for approaches to developing quality control programmes and establishing critical quality attributes for iPSC-derived medicinal products within the Eurasian Economic Union (EAEU) regulatory framework.
AIM. This study aimed to systematise global regulatory experience and EAEU regulatory requirements for the development and justification of quality control programmes for iPSC-derived medicinal products.
DISCUSSION. Medicinal products derived from iPSCs are mainly used in the treatment of neurodegenerative, cardiovascular, and oncological diseases, diabetes, graft-versus-host disease, and eye diseases. Over the past decade, specific recommendations and requirements for the quality of clinical-grade iPSCs have been published by the Chinese Society for Stem Cell Research (CSSCR), the Japanese Ministry of Health, Labour, and Welfare (MHLW), the Global Alliance for iPSC Therapy (GAiT), and the European Bank of iPSC (EBiSC). The EAEU regulatory requirements for the quality of genetically modified cells have been in effect since 2025 (Chapter 32 of Decision No. 89 of the Council of the Eurasian Economic Commission “On Approval of the Rules for Assessment of Biological Medicines in the EAEU” of November 3, 2016). The list of critical quality attributes for clinical-grade iPSCs proposed by the GAiT generally corresponds to the EAEU regulatory framework and can be used in drawing up quality control programmes for iPSC-derived medicinal products in the Russian Federation and the EAEU. Quality control programmes for finished somatic cell therapy or tissue-engineered medicinal products derived from iPSCs should be based on the principle of quality attribute traceability from the starting material onwards. The production of iPSCs is a full-fledged production process that must comply with Good Manufacturing Practice (GMP) requirements for genetically modified cells. Specific quality controls for iPSCs should include tests for residual reprogramming vector DNA, markers of the undifferentiated state, and pluripotency as part of purity characterisation, identification, and potency evaluation, respectively.
CONCLUSIONS. A quality control programme for a finished iPSC-derived medicinal product should correspond to the type of differentiated cells and take into account the indications for use. Critical quality considerations for iPSC characterisation include demonstrating the absence of contaminating undifferentiated cells or cells with new immunogenic epitopes and confirming the identity and genetic stability of iPSCs. The considered quality assessment approaches provide a basis for developing both quality control strategies for iPSC-derived medicinal products and specifications for marketing authorisation according to the EAEU requirements.
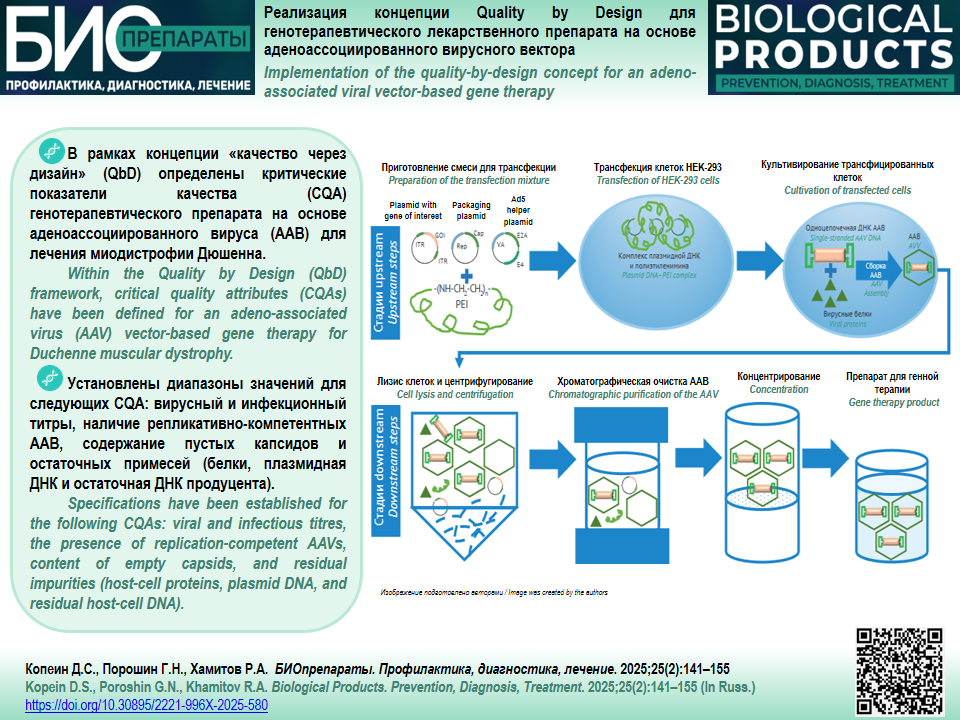
INTRODUCTION. Currently, manufacturers of adeno-associated virus (AAV)-based gene therapy products are facing a number of systemic problems stemming from the difficulties in assessing the quality of medicinal products due to insufficient scientific data, limited experience, and imperfect regulatory requirements. However, a risk-based approach to assessing critical quality attributes (CQAs) within the the framework of Quality by Design (QbD) can ensure improved efficiency in the development and production of advanced therapy medicinal products.
AIM. This study aimed to identify QbD-based CQAs and associated specifications for the development of AAV-based gene therapy products for Duchenne muscular dystrophy.
DISCUSSION. This study involved an analysis of QbD-based approaches to the development of AAV production technologies. The authors substantiated a list of the main AAV characteristics and collated available data on their impact on patients in terms of the efficacy and safety of gene therapy products and, in particular, the immune response to treatment. Following a risk assessment, the authors identified a list of CQAs for AAVs. When developing an AAV production process, the authors determined specifications for AAV CQAs, including viral and infectious titres, the presence of replication-competent AAVs, the percentage of empty capsids, and residual impurities (proteins, plasmid DNA, and residual host-cell DNA). A comprehensive risk assessment was conducted to determine the quality target product profile for an AAV-based gene therapy product for Duchenne muscular dystrophy. The authors listed the CQAs, developed the basic requirements for the applicable analytical procedures, and established the CQA specifications for the gene therapy product.
CONCLUSIONS. The use of QbD principles and risk-based approaches is an important step in CQA identification during the development of gene therapy products. The QbD methodology facilitates drafting new regulatory standards for the evaluation of the safety and efficacy of gene therapy products and helps with the development and commercial-scale manufacturing of such products.
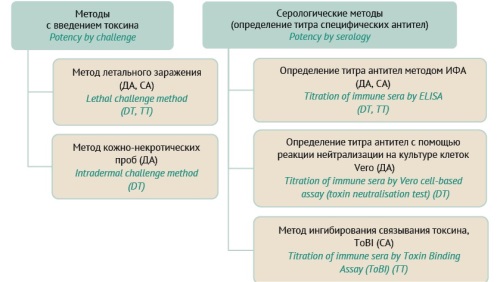
INTRODUCTION. Global vaccine manufacturers actively use modern alternative in vivo and in vitro tests to assess the potency of diphtheria and tetanus toxoids in the DPT vaccine, following 3Rs principles (replacement, reduction, refinement). At present, some in vivo tests have been completely replaced with alternative tests. In the Russian Federation, the 3Rs principles for quality assessment of DPT vaccine are practically not applied. The process of harmonization of the State Pharmacopoeia of the Russian Federation with the requirements of the regional pharmacopoeia of the Eurasian Economic Union (EAEU) necessitates the analysis of modern tests for quality assessment of DPT vaccines.
AIM. To conduct a comparative analysis of modern tests for potency determination of DPT vaccine components, assess advantages and disadvantages of tests, and identify problematic issues in harmonizing tests within the pharmacopoeia of EAEU.
DISCUSSION. In the European Union, the 3Rs principles are an integral part of the legislation. Due to investigations under the auspices of WHO and the European Directorate for the Quality of Medicines and Healthcare (EDQM), the use of laboratory animals have been completely excluded from some tests or replaced with more sparing tests that alleviate the suffering of animals. Alternative serological tests of DPT vaccine are considered sparing, but they include animals that are immunized for subsequent collection of blood samples. To establish alternative tests for potency assessment of diphtheria and tetanus toxoid, additional materials, reagents, standards, special equipment, and statistical software are required. They are multi-stage and a larger number of skilled specialists are needed. Alternative serological tests based on enzyme-linked immunosorbent assay (ELISA) have a certain advantage over the lethal challenge method: exclusion of toxins usage and determination of two types of protective antibodies in the blood serum samples obtained from one animal.
CONCLUSIONS. Despite the active development and implementation of in vitro tests for quality control of the diphtheria and tetanus components of the DPT vaccine, lethal challenge methods are more cost-effective, easy to perform and adapted to assess the direct protective effect of the vaccine. It should be noted that ELISA is the most promising test of the alternative tests applied for quality control of the DPT vaccine.
INTRODUCTION. Human hepatitis B immunoglobulins (HBIGs) are produced from the plasma of vaccinated donors and are used to prevent hepatitis B virus infection. The main challenge associated with the quality control and efficacy evaluation of HBIGs is the lack of reference standards certified for the content of antibodies to hepatitis B virus surface antigen (HBsAg). Systematisation and analysis of the relevant experience in HBIG testing and manufacturing will contribute to the development of new approaches to HBIG standardisation.
AIM. This study aimed to characterise HBIGs and consider approaches to their standardisation.
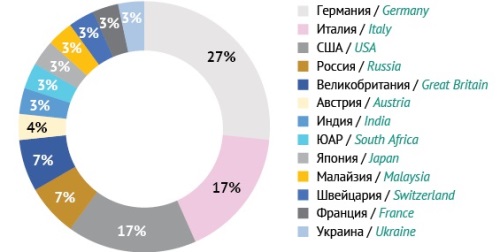
DISCUSSION. Most HBIGs are developed in Germany (27%), Italy (17%), and the USA (17%) and are formulated for intramuscular (67%), intravenous (30%), and subcutaneous (3%) administration. The potency of HBIGs varies from 50 to 500 IU/mL. There are 3 HBIGs approved in Russia, but the available HBIGs do not fully cover the needs of prevention and treatment providers. The effectiveness of hepatitis B prevention directly depends on the HBIG dose, which is calculated using the potency of the HBIG. Ensuring the accuracy of HBIG potency determination requires a reference standard for anti-HBsAg antibodies certified in international units. The existing international standard for HBIGs has limited commercial availability in the Russian Federation, and the national (pharmacopoeial) reference standard for HBIGs is lacking.
CONCLUSIONS. The analysis of information on the currently manufactured HBIGs has identified recent development trends in this pharmaceutical industry sector. The future development and implementation of the national (pharmacopoeial) reference standard for anti-HBsAg antibodies will contribute to improving the quality of HBIGs and, therefore, the effectiveness of preventive immunisation against hepatitis B.
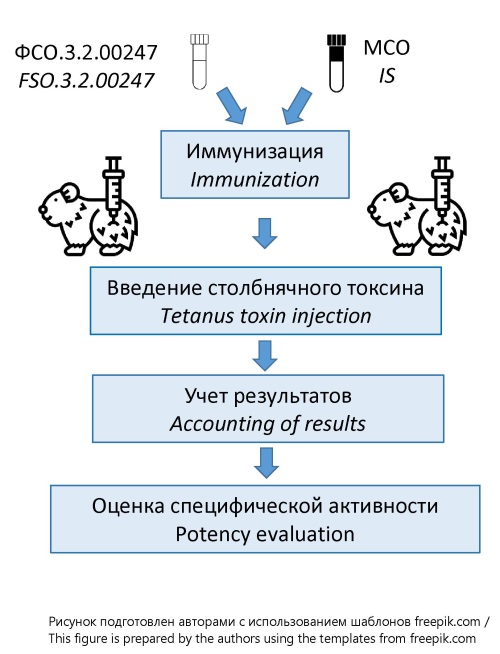
INTRODUCTION. Currently, the national pharmacopoeial reference standard (RS) FSO.3.2.00247 for the potency of adsorbed tetanus toxoid (TT) is certified only in mice, whereas the WHO International Standard (IS) for TT is certified in two animal species — mice and guinea pigs. Given the development of the Eurasian Economic Union (EAEU) regulatory framework and the harmonization of the EAEU Pharmacopoeia with the European Pharmacopoeia, it is necessary to augment the certified characteristic of FSO.3.2.00247 with the potency value determined by the lethal challenge assay in guinea pigs.
AIM. This study aimed to determine the potency of the pharmacopoeial reference standard for tetanus toxoid adsorbed using the lethal challenge assay in guinea pigs.
MATERIALS AND METHODS. The study used the 4th WHO International Standard for tetanus toxoid adsorbed and the FSO for potency testing of TT adsorbed (FSO.3.2.00247, series 011-210619). The potency of the pharmacopoeial RS was assessed relative to the IS using the lethal challenge method in guinea pigs, in accordance with the European Pharmacopoeia (monograph 2.7.8). A total of 352 outbred guinea pigs (250–350 g) were used. Animals were evenly distributed into experimental groups (8–10 animals per group) for immunization: four groups for the IS and four for the pharmacopoeial RS. After 28–30 days, the animals were injected with tetanus toxin (50 LD50). Results were evaluated over five days, with clinical signs of tetanus intoxication monitoring in accordance with the international scoring system. To control tetanus toxin activity, a group of non-immunized animals received injections of 2, 1, 0.5, and 0.25 LD50 of the toxin. The potency of the pharmacopoeial RS and the LD50 of the toxin were calculated using Kerber’s formula.
RESULTS. A certification program for FSO.3.2.00247 (series 011-210619) was developed. The pharmacopoeial RS was certified using the lethal challenge assay in guinea pigs. Four experimental studies were shown that the potency of FSO.3.2.00247 was determined to be 220 IU/ampoule. Specification of FSO.3.2.00247 (series 011-210619) may be augmented with the potency value 220 IU/ampoule for guinea pigs based on the findings obtained. The results demonstrate that pharmacopoeial RS may be used not only in the lethal challenge assay but also in alternative methods for quality control, including enzyme-linked immunosorbent assay.
CONCLUSIONS. The certified characteristic of the pharmacopoeial reference standard for potency testing of tetanus toxoid adsorbed may be augmented with the potency value determined by the lethal challenge assay in guinea pigs — 220 IU/ampoule.
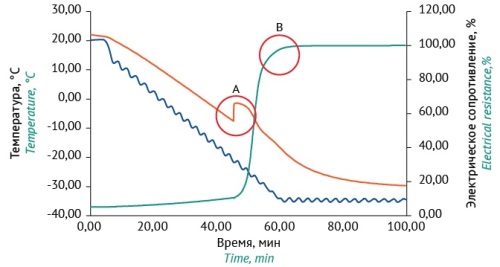
INTRODUCTION. Modern approaches to microbiological testing in the pharmaceutical industry require the use of microbial test strains standardized by the number of viable cells. The most convenient form for storage and transportation is lyophilized state, which ensures long-term preservation of the viability of microorganisms. Various drying modes for microorganisms in high concentrations (107–1012 CFU/mL) are described in the scientific literature. However, such modes cannot be directly applied while working with standardized by the number of viable cells containing 103 CFU/mL. Selection of the optimal lyophilization mode allows solving the problem of preserving the viability of microbial cells in a low concentration.
AIM. Development of a drying mode using a chamber-type apparatus that ensures the survival of microbial test strains standardized by the number of viable cells at a concentration of 103 CFU/ml.
MATERIALS AND METHODS. Microbial test strains Salmonella enterica subsp. enterica serovar Abony NCTC 6017, Staphylococcus aureus АТСС 6538 and 6538P, Alcaligenes faecalis 415, Pseudomonas aeruginosa ATCC 9027, Yersinia enterocolitica ATCC 9610, Escherichia coli ATCC 25922, Micrococcus luteus ATCC 10240 and Martin Christ Epsilon 2-4 LSCplus lyophilizer as well as sucrose-gelatin protective medium were used in the work.
RESULTS. The experiments established the optimal parameters for lyophilic drying of microorganisms at low concentrations (103 CFU/mL) in chamber-type devices: freezing to minus 25 °C, primary drying at a shelf temperature of minus 35 °C and a vacuum of 0.4 mbar for 8 h, and final drying for 4 h at a temperature of 30 °C and a residual pressure of 0.001 mbar. When using this mode, the survival rate of test strains at low concentrations ranged from 22 to 100% depending on the type of microorganism. The quality of the samples obtained was assessed using the parameter loss of drying which varied from 0.8 to 2.1%.
CONCLUSIONS. The proposed drying mode allows preserving the number of viable cells of microorganisms standardized at low concentrations using chamber-type lyophilic equipment. Freezing below the eutectic temperature may reduce survival of microbial cells after drying, as shown for A. faecalis strain 415. The survival of a set of eight microorganisms at low concentrations after lyophilization has been assessed for the first time in Russia.
PROBIOTICS
INTRODUCTION. An active search for probiotic strains of microorganisms, particularly indigenous bifidobacteria, is crucial for the development of therapeutic and prophylactic probiotics. Evaluating the probiotic potential of candidate strains requires their assessment for bacterial viability, antibiotic sensitivity, gastrointestinal stress tolerance, lysozyme resistance, and biofilm formation capacity.
AIM. This study aimed to characterise a composition of indigenous Biffdobacterium strains, B. biffdum ICIS-310 and B. longum ICIS-505, as a potential probiotic product.
MATERIALS AND METHODS. The study tested B. biffdum ICIS-310 and B. longum ICIS-505 for resistance to antibiotics, gastric acid, and bile. Mono- and co-cultures of B. biffdum ICIS-310 and B. longum ICIS-505 were tested for the number of viable cells during culture, antilysozyme activity (lysozyme resistance), and biofilm formation capacity. Antagonistic activity was tested against test strains of bacteria and fungi, including Candida albicans ATCC 24433, Proteus mirabilis ATCC 29906, Staphylococcus aureus ATCC 29213, Escherichia coli ATCC 25922, Shigella flexneri ATCC 12022, and Klebsiella pneumoniaе ICIS-278_PBV. The content of acetic acid in the culture medium was determined by gas–liquid chromatography.
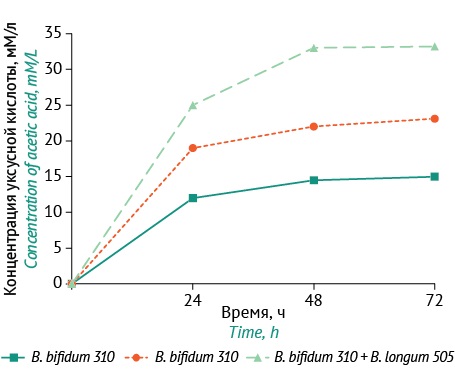
RESULTS. The indigenous strains, B. biffdum ICIS-310 and B. longum ICIS-505, were selected according to the criteria of abundance, lysozyme resistance, and biofilm formation. These strains were found to lack pathogenicity genes, exhibit resistance to a number of antimicrobials (benzylpenicillin, streptomycin, and erythromycin), and remain viable in the presence of bile for 2 hours and in the presence of gastric acid for 30 minutes. The study demonstrated the biocompatibility of Biffdobacterium cultures, with the composition of B. biffdum ICIS-310 and B. longum ICIS-505 having a higher microbial cell count after 48 hours than monocultures. When co-cultured, B. biffdum ICIS-310 and B. longum ICIS-505 demonstrated a synergistic effect, resulting in increased lysozyme resistance (up to 2.2±0.30 μg/mL×OD450), biofilm formation (up to 0.89±0.20 OD630 units), and acetate production (up to 33.2 mM/L). The antagonistic activity against test strains was more pronounced in the co-culture than in the monocultures, with the respective growth inhibition zones of 30–36 mm and 18–24 mm.
CONCLUSIONS. Indigenous B. biffdum ICIS-310 and B. longum ICIS-505 have demonstrated resistance to antimicrobial agents, bile salts, and gastric acid. Co-culturing the strains has revealed a synergistic effect on their lysozyme resistance, biofilm formation capacity, and antagonistic activity. The strains of bifidobacteria and the composition thereof hold promise for the development of novel probiotics. The evaluation of lysozyme resistance and biofilm formation capacity, as indicators of the adaptive potential of the microbiota, may be recommended for the selection and testing of probiotic strains.
DEVELOPMENT AND VALIDATION OF METHODS
INTRODUCTION. In recent years, Chikungunya virus (CHIKV) has spread in many parts of the world and has caused large-scale outbreaks with serious economic and social consequences. To improve the effectiveness of CHIKV control measures, it is necessary to develop and optimise diagnostic methods applicable not only to patient serum samples but also to mosquito samples (to identify and eliminate the foci of infection). In addition, it is important to determine antigens in culture fluid samples taken at various stages in the development and production of CHIKV vaccines.
AIM. This study aimed to develop a quantitative one-step sandwich enzyme-linked immunosorbent assay (ELISA) test system for detecting CHIKV E2 protein and a procedure for calculating the mass of whole-virion antigen in culture fluid samples.
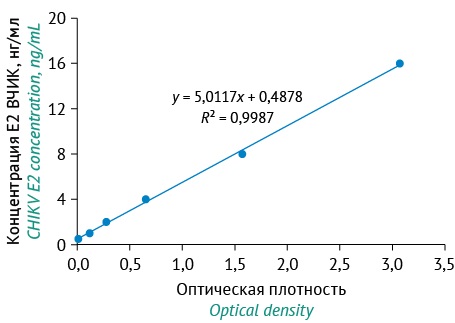
MATERIALS AND METHODS. The study focused on two mouse monoclonal antibodies, purified CHIKV (Nika21 strain, GenBank ID: PQ673601), and recombinant CHIKV E2 protein. The sensitivity of ELISA was compared with that of real-time quantitative reverse transcription polymerase chain reaction (real-time RT-qPCR). The comparison used culture fluid samples collected at different time points after infection of Vero cells with CHIKV (18, 24, 46, and 72 h). The main analytical and technical characteristics of the ELISA test system developed were determined in accordance with GOST 51352-2013.
RESULTS. The sensitivity of the assay was not less than 0.625 ng/mL, and its coefficient of variation was not more than 3.56%. The recovery of the assay was 100%. The assay demonstrated an acceptable linearity of 90–110% in the concentration range of 1.5–16 ng/mL. The specificity of the assay was 100%, as no cross-reactivity was observed with samples containing dengue, yellow fever, Sindbis, rubella, and West Nile viruses. The ELISA test system developed in this study and real-time RT-qPCR showed similar results (1.06 and 1.09 μg/mL, respectively) in calculating the mass of whole-virion antigen in culture fluid samples with the use of a conversion factor.
CONCLUSIONS. A simple, specific, and sensitive ELISA test system was developed for the quantitative determination of CHIKV E2 protein in culture fluid samples (and for rapid testing of mosquito samples). The authors offered a method for calculating the mass of whole-virion antigen from the amount of E2 protein (ELISA) and the quantity of genomic equivalents (real-time RT-qPCR). The study demonstrated a strong negative correlation between optical density values obtained using ELISA and cycle threshold values derived from real-time RT-qPCR.
INTRODUCTION. Trastuzumab is a recombinant humanised monoclonal antibody against human epidermal growth factor receptor 2 (HER2) approved as targeted therapy for patients with HER2-positive breast cancer. Improving overall patient survival, trastuzumab has become a standard of care in cancer treatment. However, trastuzumab can induce anti-drug antibodies (ADAs), which can reduce the effectiveness of treatment. Therefore, timely detection of serum ADAs is necessary for potential treatment adjustment.
AIM. This study aimed to develop and validate an enzyme-linked immunosorbent assay (ELISA) for semiquantitative determination of all-class anti-trastuzumab ADAs.
MATERIALS AND METHODS. The presented test system is a classic ELISA kit comprising a 96-well plate with trastuzumab immobilised on the inner surface of the wells, a solution of secondary antibody (trastuzumab conjugated with horseradish peroxidase), a substrate solution (3,3’,5,5’-tetramethylbenzidine), and a stop solution. The authors determined the optical density at 450 nm using a microplate photometer. Quality control solutions were prepared by diluting monoclonal antibodies against trastuzumab with a buffer containing human serum.
RESULTS. The limit of quantification for ADAs is 2 ng/mL, and the minimum required dilution is 1:2. The assay is tolerant to trastuzumab concentrations of 116 ng/mL and 1000 ng/mL and can detect ADA levels of 16 ng/mL and 100 ng/mL, respectively, in the presence of trastuzumab. The analytical procedure provides results with acceptable specificity, selectivity, and within-run and between-run precision. The assay can measure ADA concentrations ranging from 640 ng/mL to 0.16 ng/mL without exhibiting the hook effect from excess ADA levels. ADAs are stable for 6 hours at room temperature, 90 days at −70 °C, and three freeze–thaw cycles with 12-hour periods at −70 °C.
CONCLUSIONS. The test system parameters established in this study confirm the applicability of the system for detecting ADAs against trastuzumab in biological fluids during targeted therapy.

ISSN 2619-1156 (Online)































