



ОСНОВНАЯ ИНФОРМАЦИЯ О ЖУРНАЛЕ
«БИОпрепараты. Профилактика, диагностика, лечение» – научно-практический рецензируемый журнал открытого доступа, выпускаемый в печатной и онлайн-версиях, единственный в России, посвященный полному циклу разработки биологических лекарственных препаратов. Основан в 2001 году.
Цель журнала: освещение актуальных вопросов разработки регуляторных процедур, стандартизации, контроля качества, производства терапевтических, профилактических и диагностических биологических лекарственных препаратов, биомедицинских клеточных продуктов и их применения, включая клинические аспекты, для профилактики, диагностики и лечения инфекционных заболеваний, изучения аллергических и иммунопатологических процессов.
Более подробная информация – в разделе «Цели и тематика».
Целевая аудитория: биотехнологи, иммунологи, вирусологи, микробиологи, специалисты в области молекулярной и клеточной биологии, фармакологи, представители экспертных и регуляторных учреждений, специалисты в области биофармацевтической индустрии.
Учредитель и издатель: ФГБУ «Научный центр экспертизы средств медицинского применения» Министерства здравоохранения Российской Федерации.
Периодичность: 4 раза в год.
Импакт-фактор: двухлетний импакт-фактор РИНЦ (2025) – 0,641.
Редколлегия. Географическое разнообразие:
- 2 континента
- 7 стран
- 12 городов
Рецензирование:
- Двойное слепое
- Минимум 2 рецензента на рукопись
Основные метрики журнала:
- 8 дней в среднем от подачи до первого решения
- 177 день в среднем от подачи до онлайн публикации
- 31% приглашенных авторов
- 74% доля принятия рукописей
- 25 тыс. загрузок PDF в 2023 г.
Плата за публикацию: бесплатно.
Индексация. Входит в Scopus (Accepted Titles May 2025), «Белый список» научных журналов (уровень 2), Перечень ВАК (категория К1), Russian Science Citation Index (RSCI), DOAJ. Информация об индексации в других российских и международных базах доступна в разделе «Индексирование».
Регистрация. Свидетельство о регистрации средства массовой информации ПИ № ФС77-82918 от 14.03.2022 г.
Подписка. Подписной индекс в каталоге Пресса России – 57941, Урал-Пресс – 57941.
Текущий выпуск

ТЕМА НОМЕРА: РАЗРАБОТКА ПРЕПАРАТОВ ДЛЯ ЛЕЧЕНИЯ РЕДКИХ (ОРФАННЫХ) ЗАБОЛЕВАНИЙ

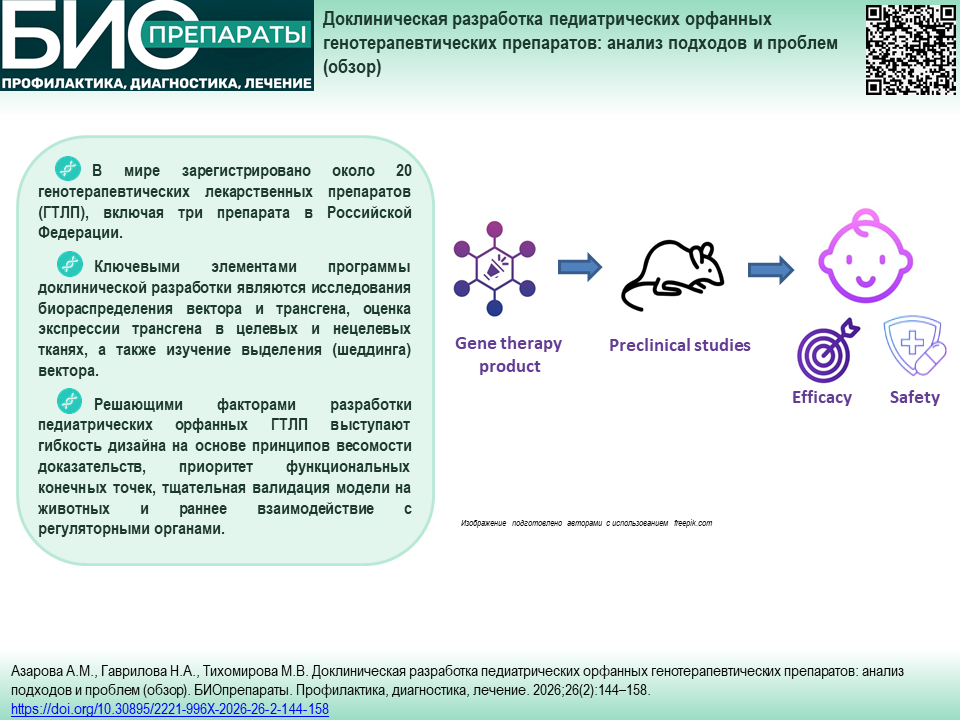

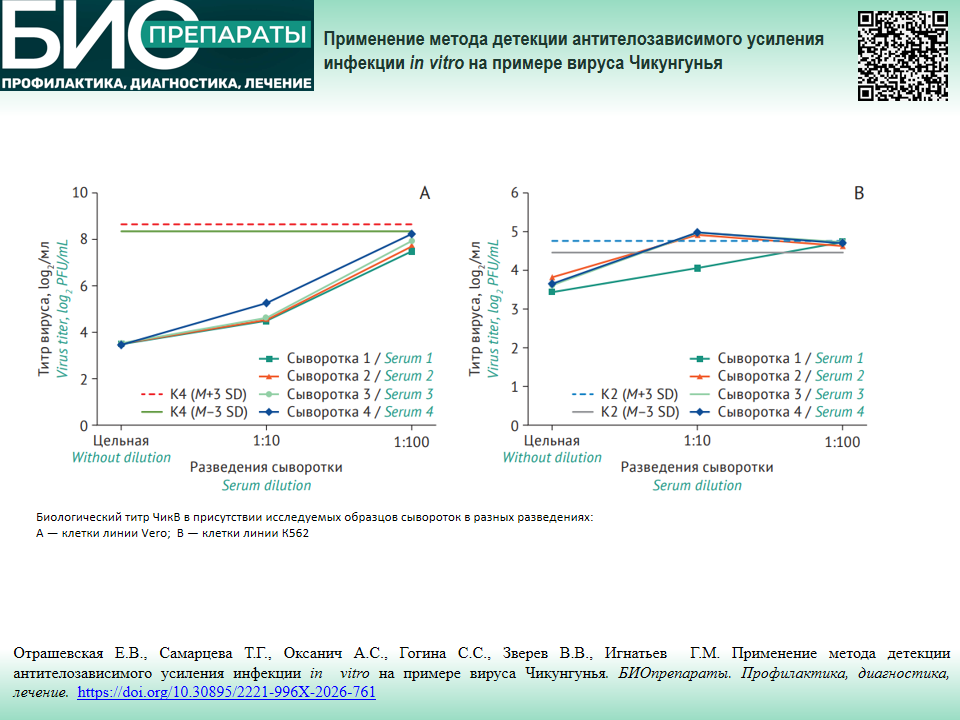
МЕХАНИЗМЫ ИММУНОПАТОЛОГИЧЕСКИХ ПРОЦЕССОВ
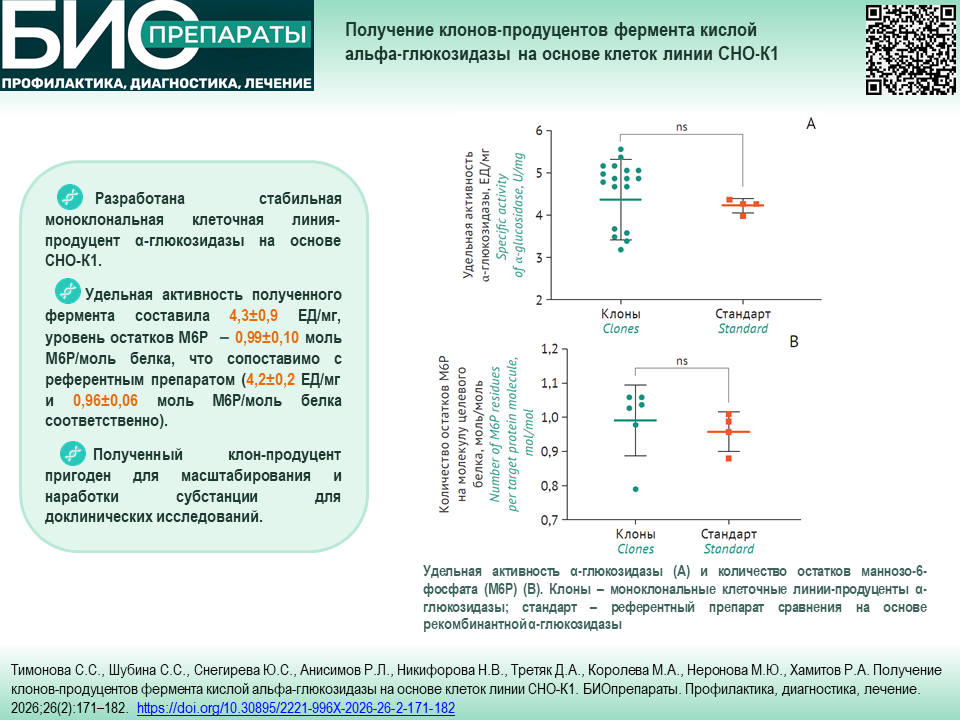
РЕКОМБИНАНТНЫЕ БЕЛКИ
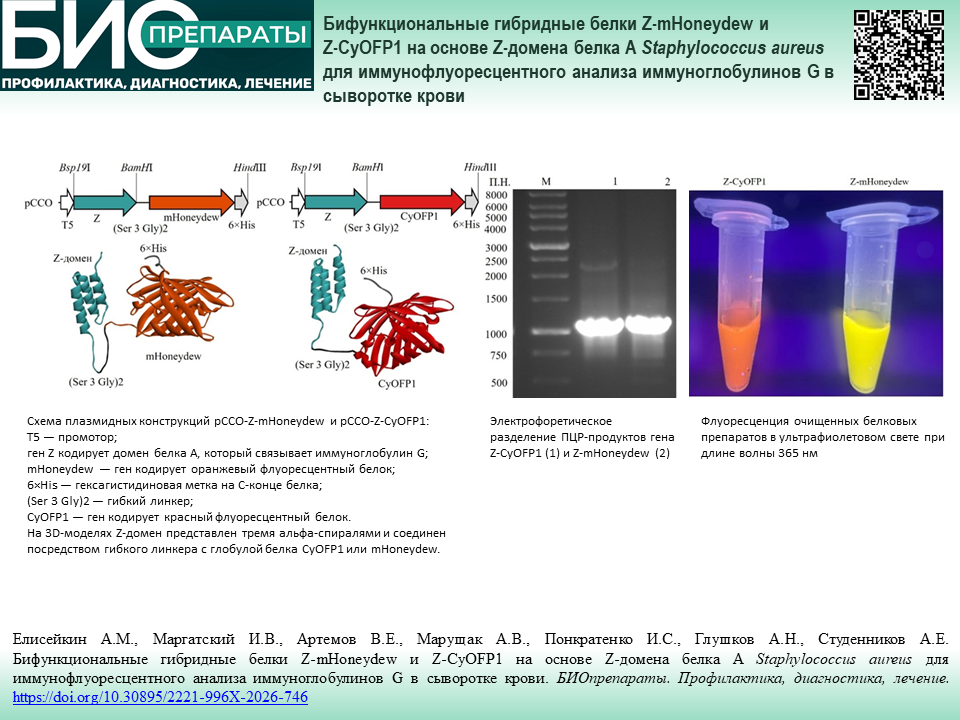
СТАНДАРТИЗАЦИЯ И КОНТРОЛЬ КАЧЕСТВА


Новости
2026-06-30
Памяти Арташеса Аваковича Мовсесянца
28 июня 2026 года ушел из жизни Арташес Авакович Мовсесянц, бывший начальник Испытательного центра экспертизы качества МИБП ФГБУ «НЦЭСМП» Минздрава России, член Государственного Фармакопейного Совета Минздрава России, член редакционной коллегии журнала «БИОпрепараты. Профилактика, диагностика, лечение».
| Еще объявления... |
































