



Issue topic INNOVATIVE BIOLOGICAL PRODUCTS: TRANSLATING FUNDAMENTAL RESEARCH INTO REAL CLINICAL PRACTICE



INTRODUCTION. High reactogenicity of the existing Ebola vaccines necessitates the development of safer and more efficacious alternatives. A solution is to use new recombinant vectorbased vaccines, such as Ad26.ZEBOV (a recombinant vaccine based on non-replicating human Ad26 serotype with an inserted glycoprotein gene of Zaire ebolavirus strain) and MVA-BN-Filo (a recombinant vaccine based on modified vaccinia Ankara strain), currently undergoing clinical trials to confirm their efficacy and safety.
AIM. This study aimed to systematize clinical trial data in order to assess efficacy, safety, and immunogenicity of the two-component recombinant Ad26.ZEBOV, MVA-BN-Filo vaccine used to prevent Ebola hemorrhagic fever in various population groups.
DISCUSSION. Literary sources were analyzed in the PubMed, ScienceDirect, and eLIBRARY.RU databases over the period of 2009–2024. Phase I–III clinical trials confirmed that the twocomponent Ebola vaccination regimen (Ad26.ZEBOV, MVA-BN-Filo) provides a strong and sustained immune response. Seroconversion reached 87–95% three weeks after the first administration and up to 100% after revaccination, with immunity lasting for at least a year. Adverse events (local and systemic, mild to moderate severity) were observed in half of the participants; serious adverse events were not associated with vaccination and occurred in less than 1%. In pregnant women, the vaccination did not increase the risk of adverse outcomes while ensuring the transmission of antibodies to the fetuses. In children and adolescents, seroconversion exceeded 90%; revaccination significantly enhanced the immune response. In HIV-infected individuals, seroconversion reached 80–85%, which is comparable to the general population. Malaria or helminthiasis did not reduce the immunization effectiveness. Considering the above, the World Health Organization and the European Medicines Agency approved this vaccination schedule in 2020 in adults and children over one year of age, including those with concomitant infections.
CONCLUSIONS. The two-component Ad26.ZEBOV, MVA-BN-Filo vaccine shows high immunogenicity and a good safety profile. The primary and booster vaccination schedule is effective in adults, children over one year of age, pregnant women, and individuals with concurrent infections, including HIV, malaria, and helminthiasis. The vaccine reduces the risk of Ebola transmission and can be used for a mass immunization strategy in endemic regions and during the disease outbreaks.
INTRODUCTION. Plasma-derived medicinal products (PDMPs) are used to treat a number of systemic diseases, immunodeficiency disorders, and life-threatening conditions. The growing PDMP consumption necessitates the scaling of production and the provision of a stable supply of plasma, with the quality meeting international standards.
AIM. This study aimed to systematize and analyze national and international compendial requirements for the quality of human plasma, including that used for PDMP production, within harmonization process of national requirements with global quality standards for the development of draft pharmacopeial monographs.
DISCUSSION. Analyzed regulatory documents and scientific literature revealed that plasma donation rates per donor ranged from 24 to 104 times per year in various countries, while acceptable donation volumes ranged from 650 to 850 mL. A two-stage donor selection system based on the results of two consecutive laboratory tests ensures high viral safety of medicinal products, which complies with international standards, including those of Plasma Protein Therapeutics Association. In the Russian Federation, the mandatory quarantine period for human blood plasma is 120 days. In the United States, plasma producers have voluntarily implemented a 60-day plasma quarantine protocol for further production. In France, plasma quarantine also lasts for two months. Analyzed viral safety control and assurance of individual donations, minipools, and fractionation pools (FPs) for viral markers in the national and regional quality standards revealed different approaches to standardization. The European and Indian Pharmacopoeias require testing of individual plasma donations for antibodies to HIV-1 and HIV-2, hepatitis C virus (HCV), and HBsAg. The US Code of Federal Regulations requires testing of individual plasma donations for HBsAg, antibodies to HIV-1, HIV-2, HCV, and HIV and HCV nucleic acids. The State Pharmacopoeia of the Russian Federation (SP RF) requires testing of each individual plasma donation for HBsAg, antibodies to HCV, HIV-1 p24 antigen, antibodies to HIV-1 and HIV-2, and the Treponema pallidum. As part of harmonizing SP RF with the monographs of foreign pharmacopoeias, requirements were established for the quality of human plasma, including that used for PDMP production.
CONCLUSIONS. The results of a comparative analysis of national and international quality requirements for fractionated plasma and virus-inactivated human plasma indicate the need to develop modern approaches to standardization and quality control. Draft pharmacopoeial monographs Human plasma for fractionation and Human virus-inactivated plasma have been prepared.
INTRODUCTION. Chikungunya virus (CHIKV) poses a major public health challenge in endemic regions due to the lack of specific preventive measures and effective antiviral therapies. Understanding and identifying viral epitopes through rigorous research directly supports the development of more effective next-generation vaccines.
AIM. This study aimed to investigate the presence of pre-existing CHIKV-specific T cell memory in unexposed individuals and identified the B cell epitopes targeted in both seropositive donors and CHIKV-infected murine models.
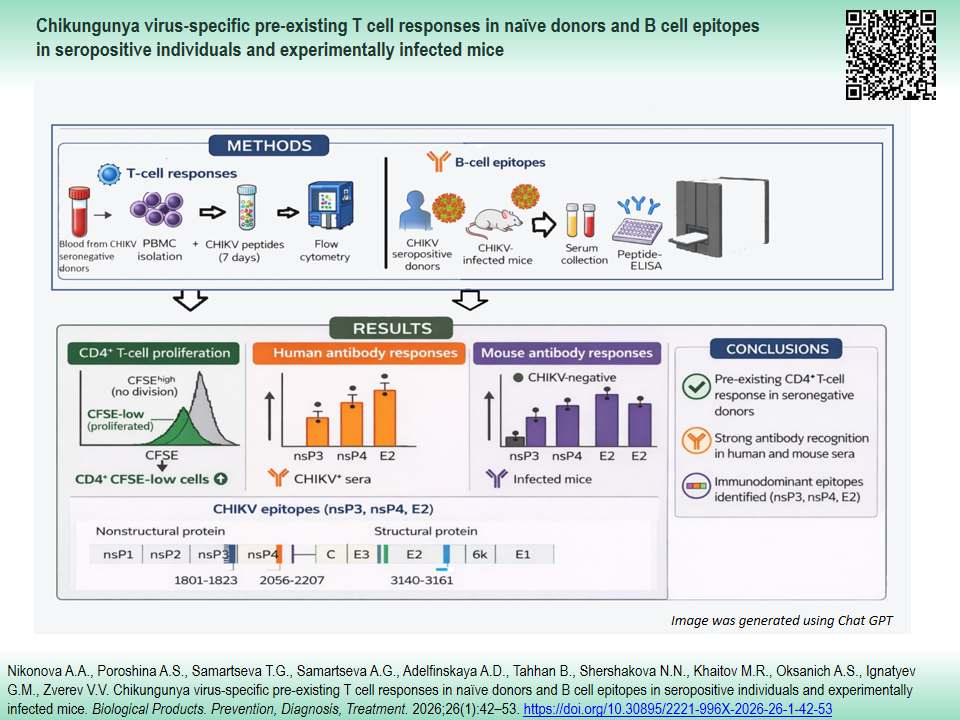
MATERIALS AND METHODS. Peripheral blood from 34 healthy volunteers and sera from four CHIKV-seropositive individuals, as well as from BALB/c mice immunized with a non-lethal CHIKV strain, were analyzed to assess Tand B-cell responses. Five synthetic peptides (21–29 aa) derived from nsP2, nsP3, nsP4, and E2 proteins were selected using the immunogenicity prediction algorithm from the Immune Epitope Database and synthesized to encompass predicted epitopes with flanking residues. Peripheral blood mononuclear cells from unexposed donors were stimulated with peptides (1.2 µg/mL), and CD4+/CD8+ T cell proliferation was evaluated by carboxyfluorescein succinimidyl ester (CFSE)-based flow cytometry. Human and mouse sera were tested for peptide-specific IgG by enzyme-linked immunosorbent assay (ELISA). Epitope localization was visualized on 3D protein models using UCSF ChimeraX software and web server I-TASSER.
RESULTS. T cell proliferation assays with CFSE labeling revealed preexisting CD4+, but not CD8+, T cell responses to peptides from nonstructural (nsP2–4) and structural (E2) CHIKV proteins in seronegative donors (P<0.05, P<0.01). Linear B cell epitopes within nsP3, nsP4, and E2 were identified by ELISA in sera from CHIKV-seropositive donors and CHIKV-infected mice. These epitopes were subsequently mapped onto 3D models of CHIKV proteins (E2, amino acids 3140–3161; nsP3, amino acids 1801–1823; nsP4, amino acids 2207–2256).
CONCLUSIONS. Our findings indicate the presence of preexisting CD4+ T cell responses in antigen-naïve individuals and underscore the importance of experimentally validating in silico–predicted epitopes for advancing serological diagnostics and vaccine development.

INTRODUCTION. Passive immunotherapy using broad-spectrum antibodies is a promising development vector of new drugs against influenza. However, production process, purification, and storage of clinically suitable recombinant antibodies still face significant challenges. Antibody scFv (single-chain variable fragments) represent a more reliable, flexible, and simpler alternative to full-length antibody analogues.
AIM. This study aimed to develop expression constructs used to synthesize scFv fragments of recombinant antibodies against influenza A and B viruses, produce scFv protein preparations, and evaluate their in vitro functional activity.
MATERIALS AND METHODS. Expression constructs encoding antibody scFv fragments were obtained by PCR using overlapping primers and genetic engineering methods. 3D models of the developed scFv fragments were constructed from the primary amino acid sequence using AlfaFold Server. Antibodies were produced in a HEK293 cell line via transient expression. Antibody preparations were purified from the culture fluid by metal affinity chromatography. Enzyme-linked immunosorbent assay (ELISA) was used to study the virus-specific activity of the antibodies. Virus neutralising activity was studied in Madine-Darbi canine kidney (MDCK) cell monolayer culture based on cytopathic effect and recorded in a haemagglutination assay.
RESULTS. Based on recombinant antibodies specific to influenza A and B viruses, configuration of scFv fragments was designed and predicted, and three genetic constructs were obtained for expression of scFv proteins in a eukaryotic cell culture. The scFv fragments were produced and purified by affinity chromatography using Ni sorbent (no less than 0.5 mg) at a concentration of about 1 mg/mL of each fragment. Protein polyacrylamide gel electrophoresis confirmed that the isolated scFv fragments matched the expected size of approximately 28 kDa. ELISA demonstrated specific binding of scFv fragments to various influenza A and B strains. It was established that a 50% virus neutralising dose of the scFv antibody fragment against the influenza virus surface haemagglutinin (170 ng/mL) is comparable to that of the original full-length antibody (179 ng/mL).
CONCLUSIONS. The scFv fragments have been designed and obtained for two antibodies; the one has broad neutralising activity against influenza A virus, the other is specific for influenza B virus. Due to their small size, scFv fragments can effectively penetrate mucous membranes upon intranasal administration. This makes scFv fragments potentially useful in the emergency prophylaxis and early therapy of acute respiratory viral infections. A promising decision for enhanced neutralising activity is to create bispecific scFv fragments capable of simultaneously targeting two viral epitopes.
INTRODUCTION. The spread of antibiotic-resistant mycobacteria necessitates new therapeutic approaches for tuberculosis, phage therapy being one of them. Liposomal mycobacteriophages are considered to be a way to enhance their efficacy; however, a comprehensive assessment of their stability is warranted for the further use of such preparations.
AIM. This study aimed to evaluate the long-term stability (over six months) of a liposomal form of mycobacteriophage D29 under storage conditions at 4 °C based on a set of physicochemical and functional parameters to substantiate its further preclinical study.
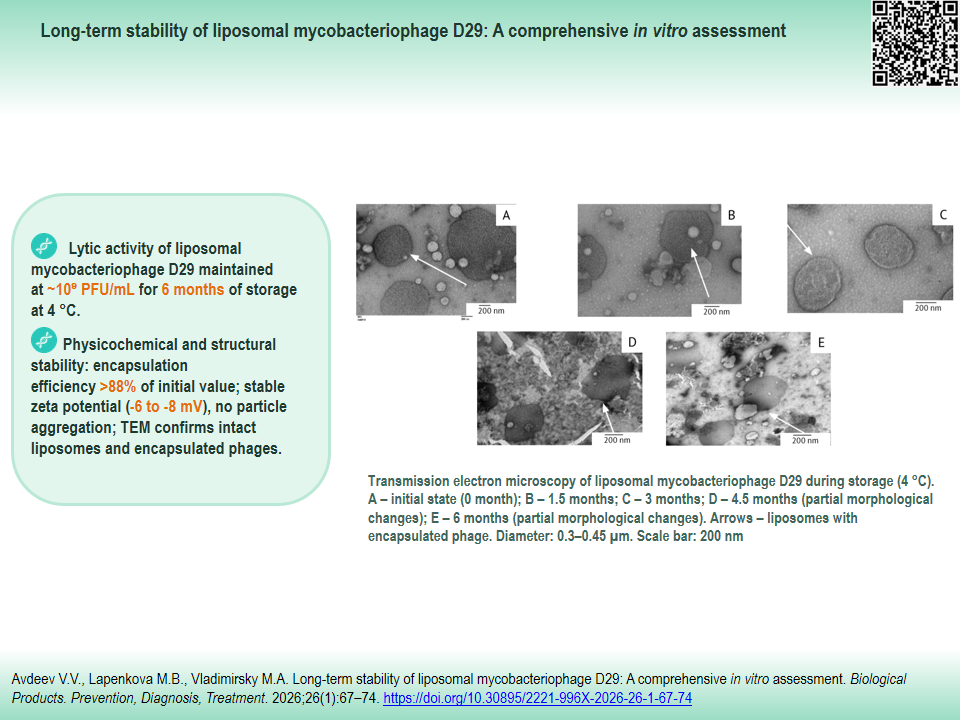
MATERIALS AND METHODS. The stability of liposomal form D29 was assessed over six months of storage at 4 °C. Sampling was performed at the following time points: 0; 1.5; 3; 4.5; and 6 months. The degree of phage encapsulation (%) was quantified by real-time polymerase chain reaction (qPCR). The surface charge (zeta potential) was measured by electrophoretic light scattering (Doppler electrophoretic mobility). Liposome morphology was analyzed using transmission electron microscopy. Functional activity was assessed by determining lytic activity via the Grazia titration method on M. smegmatis mc² 155 culture.
RESULTS. High stability of the liposomal phage D29 form was demonstrated. Lytic activity remained at a level of ~109 PFU/mL throughout the storage period (six months, 4 °C). The degree of encapsulation decreased insignificantly (>88% of the initial value). The constant zeta potential value (from –6.1±0.3 to –8.2±0.5 mV) confirmed colloidal stability of the suspension without particle aggregation throughout the entire storage period.
CONCLUSIONS. The liposomal mycobacteriophage D29 demonstrates high physicochemical and functional stability after long-term storage (six months, 4 °C), with preserved lytic activity and a high degree of encapsulation. The findings substantiate the prospects for further preclinical and subsequent clinical trial of this preparation designed to treat mycobacterial infections.
INTRODUCTION. The high variability of the measles virus RNA genome poses risks of mutations accumulating at primer binding sites (complementary binding site), potentially impacting the reliability of molecular measles diagnostics. In this study, we have assessed the impact of such mutations on the efficiency of two molecular genetic methods: PCR and loop-mediated isothermal amplification (LAMP).
AIM. This study aimed to evaluate the occurrence of mutations at primer binding sites and their impact on the efficiency of PCR and LAMP methods in detecting measles virus RNA.
MATERIALS AND METHODS. Clinical samples of nasopharyngeal secretions (LabQuest, Moscow, Russia) and nucleotide sequences from NCBI database were used in the study. AmpliTest RIBO-prep kit was used for RNA extraction. AmpliTest Measles kit and LAMP-based kit were used for amplification. MAFFT, Jalview 2.1, CD-HIT, MEGA1, and FigTree v.1.4.3 software were used to perform a bioinformatics analysis.
RESULTS. Having analyzed 1,080 nucleotide sequences of the measles virus, we found no more than one mutation in the target primers target sites and probe used for PCR. The analyzed LAMP primer binding sites showed greater variability compared to PCR primers; even in this case, 96.5% of the sequences contained no more than a single mutation at each site. Out of 69 clinical samples, only 15 had one mutation in the forward primer binding site, with a minor impact on PCR performance at low virus concentrations. For the LAMP method, three mutations were detected in the F3, B1c, and B2 primer target sites in 18 samples. In a model experiment, the mismatch in the region of the outer primer F3 did not affect the speed and sensitivity of the reaction. Inversely, the loop forming primer BIP performed abnormally well in the presence of two mutations in the RNA sequences. This can indirectly prove the existence of conformational aspects for long loop primers.
CONCLUSIONS. Both methods (PCR and LAMP) effectively detect samples with mutations; however, the LAMP method warrants further research to expand its potential for diagnosing measles.

INTRODUCTION. Filgrastim is a human granulocyte colony-stimulating factor produced using recombinant DNA technology. Since filgrastim quality control necessitates a validated method, establishing the entire method validation parameters is of high priority; confirming the previously established parameters gains primary importance when substituting critical reagents/ materials/parameters.
AIM. This study aimed to select and confirm a number of validation parameters for in vitro biological method that would assess filgrastim potency using NFS-60 cell line.
MATERIALS AND METHODS. Filgrastim potency was assessed by fluorescence intensity of alamarBlue™ reagent at excitation wavelengths of 530 nm and emission of 620 nm directly proportional to proliferation level of NFS-60 cells exposed to filgrastim 0.1 to 208 IU/mL. Statistical analysis of the results was performed using the 4-parameter logistic function 4PL and the parallel line analysis in PLA 2.0.0 software.
RESULTS. The method was deemed precise: the coefficient of variation (CV) in the repeatability study was 6.20%, and the intermediate precision study showed CV 1.19% (acceptance criterion CV≤25%). The method was linear; the coefficient of determination for the linear function was R2=0.99 (acceptance criterion R2≥0.95). The degree of recovery was 101.6% and did not exceed ±10% of the expected value. The method was robust to an increased density of the cell suspension from 1.5×105 to 3.0×105 cells/mL (CV=0.07%) and to prolonged incubation of filgrastim samples with the cell suspension (from 48 to 72 h, CV=4.2%) or with fluorescent dye (from 4 to 6 h, CV=2.05%).
CONCLUSIONS. The in vitro method assessing specific potency of filgrastim-based preparations using NFS-60 cell line is linear, precise, and accurate, and has proven to be stable under controlled changes of certain parameters. Confirmed applicability of NFS-60 cell line for assessing the potency of filgrastim-based preparations is essential for manufacturers, since it expands the range of cell cultures used for quality assessment of preparations.
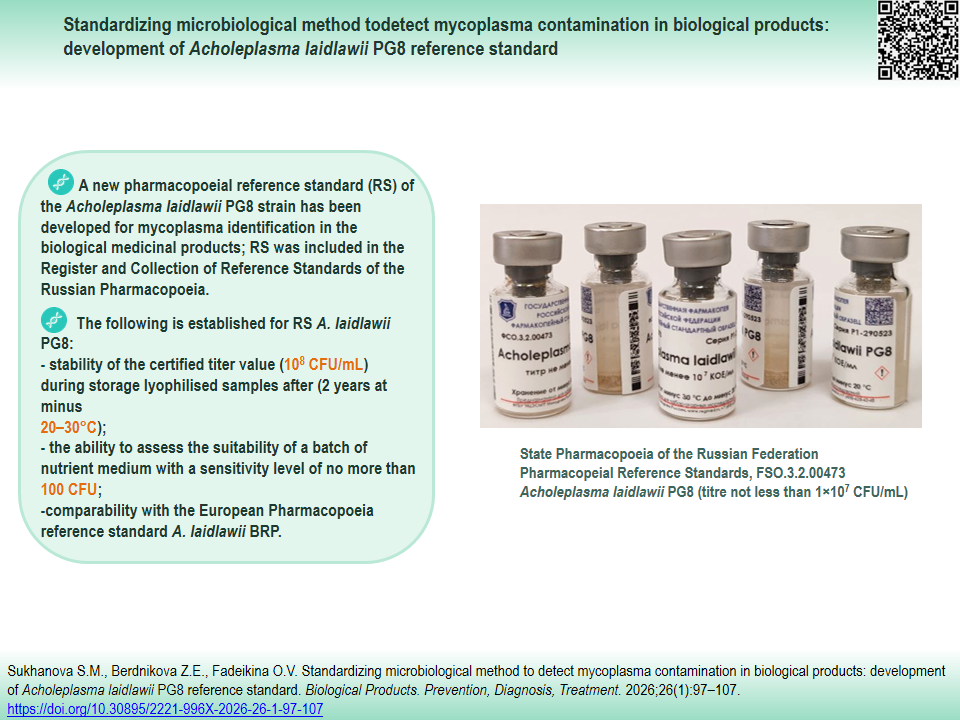
INTRODUCTION. Mycoplasmas are one of the most challenging types of microbial contamination to detect in biological products (BP). Detecting micoplasmas in the BPs and standardizing microbiological control method necessitates highly sensitive culture media and pharmacopeial reference standards (RS) for test strains of various mycoplasmas.
AIM. This study aimed to develop and certify the new pharmacopeial reference standard Acholeplasma laidlawii PG8 for mycoplasma identification in the BPs using the microbiological method.
MATERIALS AND METHODS. A. laidlawii PG8 (NCPМ 930002, ATCC 23206, NCTC 10116) strain, Kagan's medium, and horse serum was used to develop the reference standard in compliance with the State Pharmacopoeia of the Russian Federation (SP RF). The certification included determination of strain titre by the most probable number method and quality tests for: Description, Vacuum, Reconstitution time, Description of dissolved sample, Sterility, Loss on drying, and Mass homogeneity (average mass and average deviation).
RESULTS. Development and specification of A. laidlawii PG8 reference standard was completed. Lyophilised samples of the master and working bank of A. laidlawii PG8 strain culture were prepared. It was established that the lyophilization process does not significantly affect strain viability. The stability of strain titers (108 CFU/mL), cultural, and physicochemical properties of the samples was demonstrated for critical quality and reliability indicators during two years of storage at -20 to -30°C. Three batches of A. laidlawii PG8 RS were certified. The calculated titer values ranged from 10×108 to 21×108 CFU/mL. The pharmacopoeial reference standard is found to be comparable to the European Pharmacopoeia reference standard A. laidlawii BRP, batch 1 (titer 2.45×106 CFU/mL). As a result of the conducted research, A. laidlawii PG8 RS was included in the Register and Collection of Reference Standards of the Russian Pharmacopoeia.
CONCLUSIONS. A new A. laidlawii PG8 reference standard has been developed and certified that can be used to evaluate the nutritive properties of culture media; determine the inhibitory effects; and serve as a positive control in BP and material tests for the presence of mycoplasmas using the microbiological method in accordance with SP RF requirements. Introducing the new RS will contribute to standardization and improve BP control, as well as harmonization of the Russian compendial requirements with the international standards.
INTRODUCTION. The development of new effective drugs based on granulocyte-macrophage colony-stimulating factor (GM-CSF) remains a relevant objective: the study of the drug’s biological effects opens new prospects for its clinical use in the treatment of a wide range of diseases – from oncological and hematological disorders to neurodegenerative conditions. Developing a new lyophilized recombinant human GM-CSF (rhGM-CSF) based on Esсherichia coli BL21/pET-GST-6His-GM producer strain warrants an assessment of its specific activity and toxicity.
AIM. This study aimed to examine the specific proliferative activity (in vitro) and hematopoietic stimulatory effect (in vivo), as well as acute and subchronic toxicity of a new lyophilized rhGM-CSF preparation for subcutaneous administration.
MATERIALS AND METHODS. Lyophilized rhGM-CSF (strain E. coli BL21/pET-GST-6His-GM) produced by State Research Center of Virology and Biotechnology “Vector” (Russia) was used in the study. The proliferative activity was assessed in the human erythroleukemia TF-1 cell culture by XTT assay. Hematopoietic stimulatory activity was assessed in male CBA/Calac mice subjected to cyclophosphamide-induced myelosuppression (200 mg/kg, intraperitoneally). The rhGM-CSF preparation was administered at a dose of 90 μg/kg subcutaneously daily for 4 days. A leukogram was calculated on Day 5. The acute toxicity of the preparation was assessed by a single subcutaneous administration to male and female ICR mice at 500 and 1,000 μg/kg, subchronic toxicity – by four subcutaneous administrations at 90 μg/kg. On Days 1 and 7, body weight, temperature, complete blood count, biochemical markers and internal organ histology (heart, lungs, liver, kidneys, etc.) were evaluated.
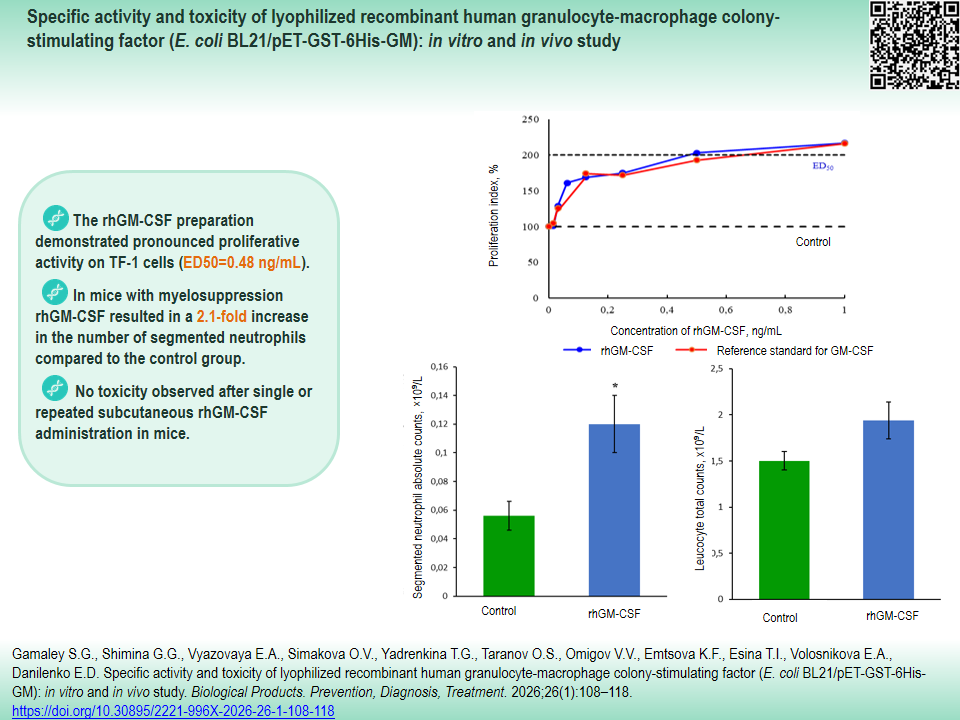
RESULTS. The rhGM-CSF preparation demonstrated pronounced proliferative activity on TF-1 cells (ED50=0.48 ng/mL). In mice with myelosuppression, the preparation resulted in a 2.1-fold increase in the number of segmented neutrophils compared to the control group. Single and repeated administration did not cause death of animals, did not affect body weight or temperature, nor did it result in significant changes of hematological or biochemical parameters or macro- and microscopic anatomy of organs.
CONCLUSIONS. The new lyophilized rhGM-CSF preparation has demonstrated high proliferative and hematopoietic stimulatory activity, as well as insignificant acute and subchronic toxicity. The obtained results substantiate further pharmaceutical development of the preparation as an effective and safe hematopoietic stimulatory agent.


ISSN 2619-1156 (Online)































