


ISSUE TOPIC: DEVELOPMENT OF MEDICINAL PRODUCTS FOR THE TREATMENT OF RARE (ORPHAN) DISEASES
INTRODUCTION. There are between 6,000 and 8,000 described rare (orphan) diseases worldwide, about 80% of which are genetic in origin and often life-threatening. Historically, the development of medicinal products (MPs) for RDs was hampered by a lack of economic incentives. Over the past 10 years, Russia has seen an active increase in the number of approved new MPs for RDs.
AIM. To trace the history of approaches to the treatment of the most common monogenic orphan diseases and analyze the problematic aspects of the development and implementation of orphan drugs in medical practice.
DISCUSSION. Historical examples of rare diseases illustrate both the slow progress of therapy from symptomatic to pathogenetic (cystic fibrosis) and the explosive growth of pathogenetic MPs of various modalities where no effective therapy existed before (spinal muscular atrophy). The path from symptomatic treatment and early replacement approaches to modern pathogenetic and etiotropic strategies is shown, including recombinant proteins, small molecules (potentiators and correctors), oligonucleotide-based drugs, enzyme replacement therapy, and gene therapy based on adeno-associated viral vectors. Special attention is paid to overcoming barriers such as viral safety, drug delivery across the blood-brain barrier ("Trojan horses"), and the challenges of commercializing gene therapy. The review examines current regulatory mechanisms and support measures for the development of orphan MPs in Russia, the EAEU countries, and the world.
CONCLUSIONS. The experience of recent decades indicates a transformation in the approach to rare diseases which have ceased to be "hopeless" and have become an area of the most intensive development in biomedical technologies. Further progress will be linked not only to the creation of new gene and cell therapy methods but also to the establishment of effective regulatory, economic, and social mechanisms ensuring equal patient access to therapy.
INTRODUCTION. The lack of uniform regulatory requirements for preclinical studies (PCS) of pediatric orphan gene therapy medicinal products (GTMPs) complicates their development and marketing authorization. This review summarizes the key features of PCS for such products and analyzes the successful experience with two registered GTMPs to support approaches for accelerating the market entry of new products.
AIM. This study aimed to critically analyze global experience in the preclinical development of GTMPs to identify key challenges in PCS and to optimize the design of studies on pediatric orphan GTMPs for expediting their translation into clinical practice.
DISCUSSION. A literature search was conducted in PubMed, Embase, Google Scholar, eLIBRARY.RU, cyberleninka.ru, and on the websites of leading regulatory agencies for the period 2020–2026. Currently, about 20 GTMPs are registered worldwide, including three products in the Russian Federation. The design of PCS for pediatric orphan GTMPs was found to be based on the weight-of-evidence principle and requires an individualized approach. Key elements of the preclinical development program include biodistribution studies of the vector and transgene, assessment of transgene expression in target and non-target tissues, and evaluation of vector shedding. The critical success factor is validation of the animal model using clinically relevant endpoints. Using Elevidys (Duchenne muscular dystrophy) as an example, the importance of assessing functional outcomes (motor activity) and correcting histological changes (reduction of muscle tissue necrosis) was demonstrated, while Zolgensma (spinal muscular atrophy) illustrated the significance of mortality reduction as the primary endpoint. The safety assessment of GTMPs is associated with the need to analyze uncertainties (long-term risks, immunogenicity), which regulatory authorities consider acceptable in the absence of alternative therapy. The obtained data confirm the necessity of early engagement between developers and regulators to optimize PCS programs.
CONCLUSIONS. The performed analysis has systematized the specific features of preclinical development of pediatric orphan GTMPs, the key ones being flexibility of design based on weight-of-evidence principles, biodistribution studies of the transgene and the vector itself, prioritization of functional endpoints, thorough validation of the animal model, and early interaction with regulatory authorities. The practical significance of the work lies in substantiating an approach that enables developers to optimize PCS programs and helps regulatory experts harmonize the assessment of efficacy and safety, thereby accelerating the entry of life-saving medicinal products into clinical practice for pediatric patients with orphan diseases.
INTRODUCTION. Artificial promoters based on the chicken beta-actin promoter (CAG, CBA, or CB) are frequently used in recombinant adeno-associated virus (rAAV)-based gene therapy products. However, data on the activity and age-related activity dynamic of these promoters remain quite limited. In this study, we quantitatively assessed CAG promoter activity in various organs of postnatal mice to determine its suitability for the development of rAAV-mediated gene therapy.
AIM. Evaluation of the relative CAG promoter activity and its dynamics in mouse organs from 3 to 12 weeks of life.
MATERIALS AND METHODS. Two-day-old ICR (CD-1) mice were injected once with rAAV9 carrying a cassette for SMN expression under the control of the CAG promoter. At 3, 6, and 12 weeks after administration, total DNA and RNA were isolated from organ samples (brain, spinal cord, liver, lungs, heart, quadriceps femoris muscle). The content of rAAV genomes and SMN mRNA in nucleic acid preparations were determined by quantitative PCR. Relative promoter activity was calculated as the ratio of the SMN mRNA concentration to the concentration of rAAV genomes, normalized on the RNA:DNA ratio in the organ. Data were analyzed using the geometric mean and regression analysis.
RESULTS. CAG promoter activity varies significantly in different organs. At week 3 after injection, the highest values were observed for the quadriceps femoris muscle, 4.7–9.7 times higher than those in other organs. It was also found that in the brain and lungs, promoter activity decreased 4.7-fold (p=0.0094) and 5.2-fold (p=0.0039), respectively, from 3 to 12 weeks after injection, while in other tissues no significant changes in activity were observed.
CONCLUSIONS. The CAG promoter is poorly suited for transgene expression in the lung and brain cells due to promoter silencing and is suboptimal for expression in the liver cells because of relatively low activity; these findings should be taken into account in the development of gene therapy products.
INTRODUCTION. The development of a biopharmaceutical drug for enzyme replacement therapy based on recombinant acid α-1,4-glucosidase (α-glucosidase) is a pressing task, the solution of which could provide patients with Pompe disease in the Russian Federation with the necessary amount of the drug. This study demonstrates an effective method for developing, from the CHO-K1 cell line, a stable industrial producer clone that produces active α-glucosidase.
AIM. This study aimed to develop monoclonal cell lines producing recombinant acid α-glucosidase and to evaluate the stability of growth characteristics and productivity during cultivation over 60 generations.
MATERIALS AND METHODS. The suspension CHO-K1 cell line (ECACC) was cultured in BalanCD Growth A medium. Cell transfection was performed by electroporation using the MaxCyte system (according to the CHO protocol). Selection of producers was carried out using puromycin (5 μg/mL). Cloning was performed using a cell dispensing system based on microfluidic technology (C.SIGHT). The monoclonality of the cell lines was confirmed using an automated cell imaging system (Cell Metric CLD). The concentration of α-glucosidase in the culture fluid was determined by enzyme-linked immunosorbent assay. Enzyme activity was measured by a colorimetric method using the substrate 4-nitrophenyl-α-D-glucopyranoside (pNP-α-D-Glc).
RESULTS. Screening of 1000 producers was performed, resulting in the selection of 22 cell lines with a productivity of 0.14–0.65 g/L. The lead α-glucosidase producer GAA-14 was cloned, followed by confirmation of monoclonality. A panel of 20 monoclonal lines was obtained. The specific activity of the enzyme and the mannose-6-phosphate residue (М6Р) content did not differ from those of the reference drug (4.3±0.9 U/mg and 0.99±0.10 mol M6P/mol protein, respectively). When studying the stability of the lead clone over 60 generations, preservation of growth characteristics (viability, 98.5±1.5%; population doubling time, 20.0±1.3 h; and productivity, 450±20 mg/L) was demonstrated under batch cultivation over 7 days.
CONCLUSIONS. A stable monoclonal α-glucosidase producer cell line based on CHO-K1 cells has been developed, yielding the active enzyme at 0.45 g/L on day 7. The resulting producer clone is suitable for scale-up and manufacture of the drug substance for preclinical studies.
MECHANISMS OF IMMUNOPATHOLOGICAL PROCESSES
INTRODUCTION. The mechanism of antibody-dependent enhancement (ADE) caused by the Fcγ receptor on immune cells is a key factor. When the virus binds to specific immunoglobulin G
(IgG), it enhances cell penetration. ADE can also occur in vaccinated individuals, making its study crucial for evaluating vaccine safety.
AIM. This study aimed to apply the method of detection of antibody-dependent enhacement in vitro using cells of the K562 and Vero lines, as well as the Chikungunya virus strain Nika21.
MATERIALS AND METHODS. Chikungunya virus (ChikV) Nika21 strain and different dilutions sera containing anti-ChikV IgG were used. To evaluate ADE, K562 and Vero cells were challenged with a ChikV with specific IgG mixture. On the fourth day after infection, the biological ChikV titer and viral RNA content were determined. ChikV titer was determined by standard in vitro virus titration and RNA amount was determined by real-time fluorescence reverse transcription PCR. ADE was considered confirmed if the values of the biological ChikV titer in the presence of the tested serum were >3 SD relative to the mean ChikV titer in the control, which represented ChikV with serum of a healthy volunteer.
RESULTS. In Vero cells, both methods showed increased ChikV replication with an increase in the dilution of immune sera, which, however, did not exceed the values of ChikV titers in the control. In K562 cells, serum with anti-ChikV IgG in one dilution demonstrated statistically significant enhancement of ChikV replication relative to the corresponding control. Thus, in our study, ADE was detected in the presence of intermediate dilutions of immune sera with a relatively medium or low titer of anti-ChikV AT.
CONCLUSION. ChikV replication in FcR-negative Vero cell line confirmed the high sensitivity to the virus and the possibility of using it exclusively for assessing the neutralizing activity of specific IgG. FcR-expressing K562 cell line allowed to analyze both virus neutralizing and activating infection functions of IgG. Our results confirmed the possibility of conducting ADE studies in vitro, which greatly simplifies the study of the ADE phenomenon when assessing the safety of vaccines under development.
RECOMBINANT PROTEINS
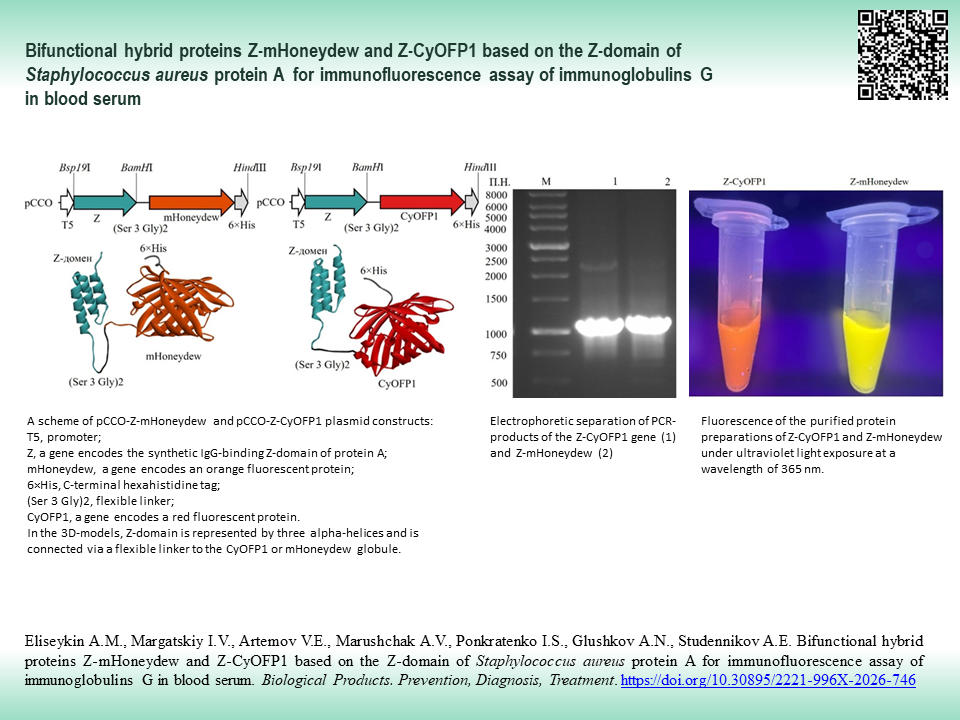
INTRODUCTION. Traditional methods for detecting serum IgG rely on antibodies chemically conjugated to enzymatic or fluorescent tags. The efficiency of such methods may be limited by steric hindrance during chemical cross-linking. As an alternative, bifunctional hybrid proteins Z-mHoneydew and Z-CyOFP1, based on the Z-domain of Staphylococcus aureus protein A and bright fluorescent proteins mHoneydew and CyOFP1, combining the binding to IgG and intense fluorescence, have been proposed for IgG immunofluorescence analysis; their production excludes the stage of chemical conjugation.
AIM. This study aimed to produce hybrid proteins based on the Z-domain of protein A for the creation of universal fluorescent reagents for the detection of IgG in blood serum.
MATERIALS AND METHODS. The pCCO-Z-mHoneydew and pCCO-Z-CyOFP1 genetic constructs were created by molecular cloning in the pCCO vector. Z-mHoneydew and Z-CyOFP1 proteins (~35 kDa) were expressed in Escherichia coli strain M15. Protein purification was performed by affinity chromatography on Ni²⁺-NTA agarose. The protein properties were studied using spectrofluorimetry, enzyme-linked immunosorbent assay (ELISA), and immunofluorescence assay.
RESULTS. Genetic constructs pCCO-Z-mHoneydew and pCCO-Z-CyOFP1 under the control of the T5 promoter encode chimeric proteins with the following structure: Z-domain of protein A — flexible linker (Ser-Ser-Ser-Gly-Ser-Ser-Ser-Gly) — fluorescent protein mHoneydew or CyOFP1 — 6×His tag. Soluble proteins were obtained with a yield of ~37 mg/L (Z-mHoneydew) and ~6 mg/L (Z-CyOFP1) with a purity ≥90%. For the Z-mHoneydew protein, excitation and emission maxima were observed at 480 and 536 nm, respectively; a 26 nm blue shift in emission was observed relative to native mHoneydew (562 nm). For Z-CyOFP1, the excitation maximum was recorded at 515 nm; an 18 nm red shift in excitation was observed relative to native CyOFP1 (497 nm), while maintaining the emission maximum. Dose-dependent binding of IgG to the hybrid proteins was demonstrated using ELISA. A linear dependence was observed for Z-mHoneydew in the concentration range of 30–235 ng/μL (R²=0.96) and 15–250 ng/μL (R²=0.97) for Z-CyOFP1, confirming the capacity of the hybrid proteins to bind IgG. Immunofluorescence assay showed the feasibility of detecting IgG, specific to conjugate benzo[a]pyrene-bovine serum albumin, in rabbit serum using hybrid proteins. A linear relationship was observed for Z-mHoneydew in the serum dilution range of 1:10–1:160 (R²=0.92) and 1:10–1:80 (R²=0.98) for Z-CyOFP1, evidencing the suitability of hybrid proteins for detection of specific antibodies in blood serum.
CONCLUSIONS. Hybrid proteins Z-mHoneydew and Z-CyOFP1 retain fluorescent properties and the capacity to bind to IgG, including serum IgG, which is promising from the point of view of developing prototypes of universal immunofluorescent test systems.
QUALITY CONTROL AND STANDARDISATION
INTRODUCTION. The degree of adsorption of vaccine components on the adjuvant determines vaccine efficacy. Requirements for the degree of adsorption of antigen in the finished product have not been established by the WHO, whereas they are regulated in the Russian State pharmacopoeia. These differences in requirements limit the comparable quality assessment of DTP vaccines from different manufacturers, and the lack of international standards complicates adequately assessing the quality of a wide variety of vaccines.
AIM. To analyze regulatory requirements and literature data for determining the completeness of adsorption of diphtheria and tetanus toxoids in combined vaccines in order to improve quality control systems and harmonize national requirements with global quality standards.
DISCUSSION. Currently, there are no international requirements for the quality of aluminum hydroxide gel for adsorption or for the degree of adsorption of components of DTP vaccine. Leading global pharmacopoeias establish quality standards for aluminum hydroxide gel used for adsorption; however, no requirements have been developed in the Russian Federation. Manufacturers independently establish requirements for the degree of adsorption of antigen for each product, determining this value at least at the stage of obtaining the adsorbed antigen. This quality indicator may not be included in the specifications for the finished product. There are three main methods for assessing the degree of adsorption. They determine the amount of unbound antigen in the supernatant; their sensitivity and cost vary. Antigen content is expressed in generally accepted flocculation units — Lf (Limit of Flocculation). Domestic vaccines differ from foreign ones in the methods for determining the degree of antigen adsorption and the units used to express the quantitative content of tetanus toxoid. Due to the differences in approaches and units for expressing the degree of adsorption, domestic vaccines are inferior to foreign ones. However, this fact does not indicate the low efficacy of domestic DTP-based vaccines.
CONCLUSIONS. Standardizing aluminum hydroxide gel quality requirements for adsorption, antigen adsorption monitoring methods, and units for expressing quantitative antigen content is crucial for harmonizing pharmacopoeial requirements. Implementing current adjuvant requirements and current antigen adsorption quality control methods will simplify regulatory frameworks for DTP-based vaccines.
INTRODUCTION. The assessment of allergen activity by competitive enzyme-linked immunosorbent assay (ELISA) requires reference standards (RS) of specific sera containing IgE antibodies to the common ragweed pollen allergen. However, neither an international nor a national standard for such serum is available. Therefore, the development of a Russian pharmacopoeial reference standard (PhRS) for IgE-containing serum to common ragweed pollen allergen is relevant.
AIM. This study aimed to certify a candidate for the pharmacopoeial reference standard of lyophilized IgE-containing serum to common ragweed pollen allergen (PhRS IgE-Amb) and to evaluate the feasibility of its use for determining IgE concentration and allergen activity.
MATERIALS AND METHODS. The concentration of allergen-specific IgE antibodies in the candidate for the PhRS IgE-Amb was determined by reverse allergosorbent test (REAST) using a commercial reagent kit manufactured in Russia. The identity of the candidate for the PhRS IgE-Amb was determined according to General Pharmacopoeia Monograph 1.7.2.0034.15 by direct ELISA. Tests of the candidate for the PhRS IgE-Amb for the quality attributes (Description, Water, Mass homogeneity, Solubility, and Tightness) were conducted in accordance with the requirements of the State Pharmacopoeia of the Russian Federation (SP RF). Allergenic activity was assessed by competitive ELISA. Statistical analysis was performed using analysis of variance (ANOVA).
RESULTS. The candidate for the PhRS IgE-Amb was certified according to the approved certification program. The certified characteristic of the PhRS IgE-Amb, i.e., the concentration of specific IgE antibodies to common ragweed pollen allergen, was determined by the REAST method and found to be 27.02±4.6 IU/mL (class 4). The specificity of binding of the PhRS IgE-Amb to common ragweed pollen allergen was confirmed. The results of tests for additional quality attributes (Description, Water, Mass homogeneity, Solubility, Tightness) met the requirements of the SP RF. The feasibility of using the PhRS IgE-Amb for determining the concentration of IgE antibodies in enterprise reference sera by direct ELISA and for assessing common ragweed pollen allergen activity by competitive ELISA was demonstrated. The coefficients of variation between the known values and those determined using the RS were 4.7% for direct ELISA and 8.0% for competitive ELISA.
CONCLUSIONS. For the first time, a Russian pharmacopoeial reference standard of lyophilized IgE-containing serum to common ragweed pollen allergen has been certified with the established characteristic — the concentration of specific IgE antibodies to common ragweed pollen allergen determined by REAST. The PhRS IgE-Amb complies with the requirements of the SP RF, is recommended for inclusion in the State Register of Reference Standards, and enables the standardization of allergens and reference sera in the Russian Federation.
INTRODUCTION. The reactogenicity of whole-cell pertussis vaccines (wPV) is largely attributable to the presence of lipooligosaccharide (LOS), a bacterial endotoxin (BE) from the outer membrane of Bordetella pertussis. Assessing the LOS content in wPV and its correlation with specific safety parameters would help establish an acceptable LOS level that does not increase reactogenicity.
AIM. This study aimed to determine the lipooligosaccharide content in whole-cell pertussis vaccines and establish its correlation with specific safety parameters in mice (body weight change and specific safety index).
MATERIALS AND METHODS. Experimental batches of wPV manufactured from production strains (38, 475, 703) and from B. pertussis circulating strains (1-20, 2-20, 16-16, 25-16, 28(1)-18, 30-18, 33-18), isolated from children with pertussis, were studied. BE content was measured using the gel-clot test and the turbidimetric method. Specific safety parameters were assessed 24 hours and 7 days after administration by monitoring body weight changes in outbred mice of both sexes and by calculating the specific safety index (%).
RESULTS. BE content was determined at three time intervals: within 1 year after wPV manufacture (prior to combining with other diphtheria, tetanus, and pertussis (DTP) vaccine components); from 1 to 2.5 years (shelf life of wPV as part of the DTP vaccine); and from 2.5 to 6 years (after expiry). Mean BE content in wPV from circulating strains stored for 1 year and for 1–2.5 years ranged from 41,872.9 to 70,576.0 EU/mL; in wPV from the production strain stored for 1–2.5 years, it was 43,980.6 EU/mL. Vaccines stored for 2.5–6 years showed mean BE values ranging from 1353.8 to 26,655.0 EU/mL. A wide range of BE content was observed across manufactured batches, varying from <1000–1270 EU/mL (strain 30-18) to 36,430–94,270 EU/mL (strain 703). Specific safety index values for all samples, except for the freshly prepared 16-16 and 33-18, were >60%. A moderate inverse correlation was found between BE content and both mouse body weight change and the specific safety index.
CONCLUSIONS. BE content did not differ statistically between wPV samples stored for 1 year and those stored for 1–2.5 years. The specific safety index values (>60%) indicate that the BE levels comply with safety requirements. The inverse correlation between BE content (in vitro) and the specific safety index (in vivo) confirms the role of LOS in the specific toxicity of the vaccine, supporting further investigation into its levels in B. pertussis strains and its impact on wPV safety.

ISSN 2619-1156 (Online)































