



ISSUE TOPIC: VACCINES: CLINICAL TRIALS AND ACTUAL CLINICAL PRACTICE
INTRODUCTION. The development of universal influenza vaccines based on conserved influenza virus antigens is a promising strategy for preventing pandemic influenza. Recombinant vaccines based on adenoviral vectors have high antiviral potential and have proven their effectiveness during the COVID-19 pandemic. In this regard, the development and clinical evaluation of a viral vector vaccine against influenza A seem relevant.
AIM. The aim was to evaluate the safety, reactogenicity, and immunogenicity of a broad-spectrum viral vector vaccine against influenza A after a single intranasal administration in healthy volunteers.
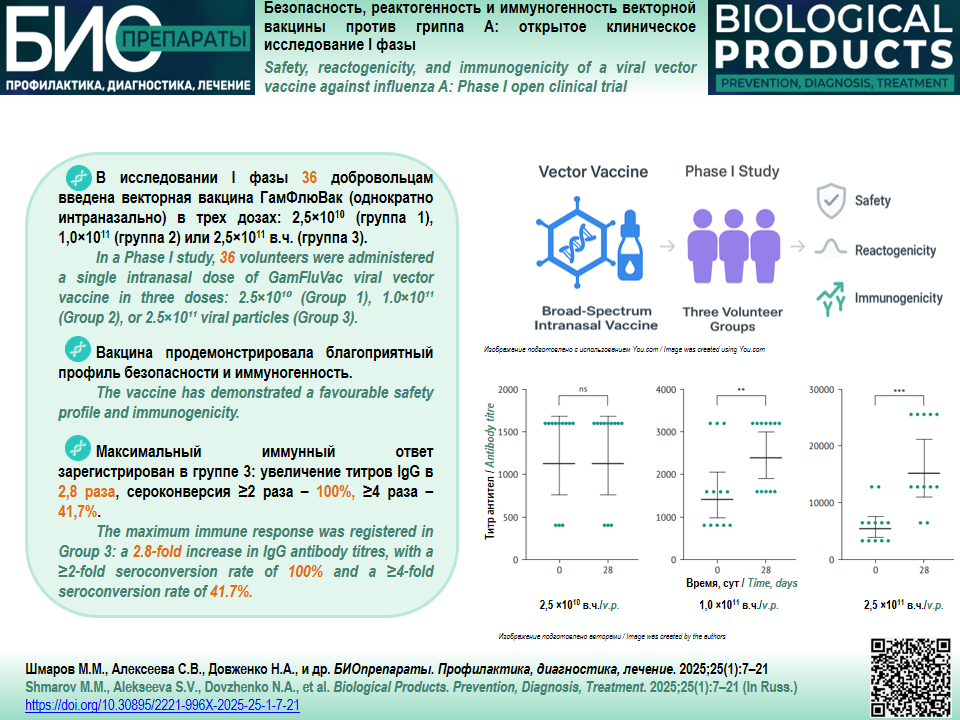
MATERIALS AND METHODS. This clinical trial studied the GamFluVac recombinant human adenovirus serotype 5 (rAd5)-based vaccine against influenza A (National Research Center for Epidemiology and Microbiology named after the honorary academician N.F. Gamaleya). The clinical trial enrolled 36 volunteers. The follow-up period was 28 days. The safety assessment of the viral vector vaccine was based on the incidence, nature, and severity of adverse events (AEs) after a single intranasal administration at doses of 2.5×1010 (Group 1), 1.0×1011 (Group 2), and 2.5×1011 (Group 3) viral particles. Immunogenicity was evaluated by measuring serum IgG antibodies against influenza A (H5N2) by enzyme immunoassay on Day 0 and Day 28.
RESULTS. No serious or severe AEs were reported during the clinical trial. Most AEs associated with the vaccine manifested as respiratory disorders, abnormal blood findings, and general disorders (elevated body temperature, headache, chills, malaise, arthralgia, and myalgia). Statistically significant differences (p=0.0285) were identified in the incidence of general disorders and administration site conditions in Group 1 (0%), Group 2 (16.7%), and Group 3 (33.3%). Group 3 demonstrated the highest increase in the geometric mean titres of specific IgG antibodies (2.8 times the baseline) on Day 28. In this group, 100% of volunteers had a ≥2-fold seroconversion rate, and 41.7% of volunteers had a ≥4-fold seroconversion rate.
CONCLUSIONS. This phase I clinical trial of the GamFluVac viral vector vaccine against influenza A demonstrated the immunogenicity and favourable safety profile of the vaccine after a single intranasal administration.
INTRODUCTION. Currently used inactivated vaccines with acellular/whole-cell pertussis components do not provide sterilising or sufficiently long-lasting immunity, nor do these vaccines prevent the spread of the pathogen or infection. A live pertussis vaccine for intranasal administration, GamLPV, has passed the necessary preclinical studies and a clinical trial that established the optimal dose for single-dose vaccination.
AIM. This study aimed to optimise the method and schedule of administration of the GamLPV intranasal live vaccine for pertussis prevention on the basis of the immunogenicity results obtained in a clinical trial involving healthy volunteers aged 18 to 40 years.
MATERIALS AND METHODS. This blind, randomised, placebo-controlled clinical trial enrolled 50 healthy adults, who were then randomised into two groups according to the method of GamLPV administration (drops or spray). The study evaluated the efficacy of two administration schedules, including single-dose vaccination and double-dose vaccination with an interval of 60 days. The study analysed nasopharyngeal and oropharyngeal aspirates, serum samples, and peripheral blood mononuclear cells from volunteers. The levels of IgM, IgA, and IgG antibodies specific to Bordetella pertussis and the levels of secretory IgA antibodies were measured in serum samples and nasal aspirates, respectively, by enzyme-linked immunosorbent assay. Agglutinating antibody titres were determined by agglutination tests. The quantitative determination of B. pertussis DNA in aspirates used real-time polymerase chain reaction. Cell-mediated immune responses were assessed by the production of IFN-γ and IL-17 cytokines in peripheral blood mononuclear cells.
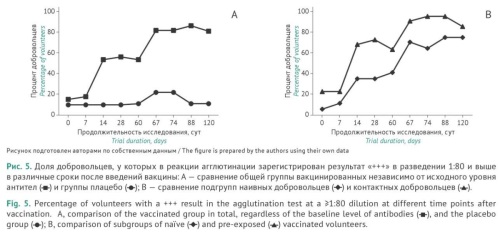
RESULTS. GamLPV administration to volunteers induced B. pertussis-specific IgG and IgA antibodies observed in serum samples, secretory IgA antibodies identified in aspirates, and increased titres of total antibodies to B. pertussis measured by agglutination tests, regardless of the vaccination schedule and the method of administration. Repeated GamLPV administration had a pronounced booster effect, as evidenced by elevated titres of antibodies of all classes from day 14. Moreover, repeated vaccination reduced the time for clearance of B. pertussis bacteria compared with that after the first vaccination (from 28 to 14 days). There were no significant differences in the time courses and values of the measured parameters for nasal vaccine delivery by spray and drops.
CONCLUSIONS. Intranasal vaccination with GamLPV induces pronounced humoral and cellular immune responses and nasal mucosal immunity against pertussis infection in adults. The recommended method for GamLPV administration is nasal drops. The accelerated clearance of bacteria observed after the second vaccination is indicative of sterilising immunity provided by the first vaccination, which can ultimately reduce pertussis transmission in the population.

INTRODUCTION. Chickenpox is a highly contagious viral disease that primarily affects preschool children. A number of chickenpox vaccines are licensed worldwide, but there are still gaps in vaccination coverage. Chickenpox vaccines may differ in efficacy, and certain issues with their long-term effectiveness remain unresolved. Chickenpox vaccines may have different safety profiles, and there are lingering concerns about adverse effects. These considerations highlight the need for further safety monitoring and the development of vaccination programmes.
AIM. This study aimed to summarise Russian and international experience in studying the safety, efficacy, and effectiveness of chickenpox vaccines to improve and optimise immunisation strategies.
DISCUSSION. This paper presents an analysis of information on the development of chickenpox vaccines and the assessment of their safety, efficacy, and effectiveness based on clinical trial results and real-world evidence. Most licensed vaccines are produced from the original Oka strain of chickenpox virus (Varicella zoster), while a South Korean company produces a vaccine using its own MAV/06 strain. Chickenpox vaccines manufactured in Belgium, the USA, China, and South Korea have demonstrated comparable safety, immunogenicity, efficacy, and effectiveness. Regional immunisation programmes have significantly reduced chickenpox incidence and complications, and the inclusion of chickenpox vaccination in the national immunisation schedule of the Russian Federation is anticipated in the foreseeable future. The national immunisation programme may include vaccines that have been properly studied.
CONCLUSIONS. According to the analysis of national and international experience, live attenuated vaccines have comparable efficacy, effectiveness, and immunogenicity and are safe for human use. Consequently, chickenpox vaccines can be used in the development of the national immunisation programme in the Russian Federation.

INTRODUCTION. Cases of tetanus are registered annually throughout the world, mainly in unimmunised or incompletely immunised populations. Analysis of tetanus cases and identification of the reasons for non-vaccination, including refusal to vaccinate, are important for drawing the attention of health professionals to this issue.
AIM. This study aimed to review case reports of tetanus in unvaccinated or partially vaccinated individuals, analyse reasons for non-vaccination, and identify problems associated with preventive vaccination against tetanus.
DISCUSSION. According to epidemiological data, cases of tetanus are recorded every year in almost every country in the world. In 2023, the World Health Organisation (WHO) reported 21,830 cases of tetanus worldwide, and the Russian Federation reported 8 tetanus patients, including children. The main issue with diagnosing tetanus lies in the lack of reliable laboratory tests for confirming tetanus. Tetanus-specific therapy with tetanus antitoxin (equine) is associated with the risk of allergic reactions. Traditionally, tetanus is considered an infection that develops only in patients with deep and soil-contaminated wounds. However, unvaccinated or partially vaccinated individuals are at high risk of tetanus, even with minor wounds. This study involved an analysis of case reports of tetanus (13 cases) in unvaccinated or partially vaccinated individuals with minor wounds or wounds minimally contaminated with soil. In all the paediatric tetanus cases discussed in this article, the parents had not vaccinated their children for religious and/or personal reasons. The analysis of case reports of tetanus in adults showed that the patients had not taken their wounds seriously and had not sought medical help before the onset of the disease.
CONCLUSIONS. The concerning incidence of tetanus is attributed to insufficient public awareness of the dangers of the disease and the rising number of people refusing vaccines. Health professionals, public organisations, and religious communities should work together to promote vaccination and improve health education. This will enhance public confidence in vaccination, increase preventive vaccination coverage, and reduce the incidence of tetanus.
BIOTECHNOLOGICAL MEDICINAL PRODUCTS

INTRODUCTION. Currently, the primary treatment method for botulism is the use of botulinum antitoxin, which causes a number of side effects, including allergic reactions. The development of medicinal products based on monoclonal antibodies (mAbs), in particular, single-domain mAbs fused to the human IgG1 Fc fragment, holds promise for the treatment of botulinum toxin poisoning.
AIM. This study aimed to optimise the technology for laboratory-scale production of a single-domain mAb fused to the human IgG1 Fc fragment (B11-Fc) for botulism treatment and post-exposure prophylaxis and to conduct a preclinical efficacy study of this mAb.
MATERIALS AND METHODS. The study used CHO cells. B7, a stable clone producing the B11-Fc single-domain mAb, was cultured in Erlenmeyer flasks using commercially available media and feeds. The B11-Fc mAb was purified using multistep chromatography (including affinity, anion exchange, and multimodal chromatography steps), virus elimination, and tangential flow filtration. The purity of the B11-Fc mAb was assessed by high-performance liquid chromatography (HPLC) and electrophoresis. The glycan profile was established by HPLC. Bio-layer interferometry was used to measure the mAb concentration in the culture fluid and to determine the equilibrium dissociation constants for the mAb and various Fc receptors. Botulinum toxin type A (BoNT/A) was produced by culturing the Clostridium botulinum A98 strain and purified by chromatography. In vivo experiments involved intraperitoneal and intragastric administration of BoNT/A to female BALB/c mice, with a subsequent assessment of the severity of toxic signs. The B11-Fc mAb was administered intramuscularly or intravenously (to study the pharmacokinetics). The efficacy of the B11-Fc mAb (in terms of mouse survival) was studied using various toxicity models and the prophylactic and therapeutic modes of administration.
RESULTS. The study optimised culture conditions for the B11-Fc mAb producer clone and developed a mAb purification technology that ensured a high yield (0.5 g/L) and a purity of over 99%. The average particle size in the mAb preparation was 7.85 nm. The study characterised the glycan profile of the B11-Fc mAb and determined the equilibrium dissociation constants for the mAb and human Fc receptors. Poisoning with BoNT/A was modelled in mice. The intramuscular administration of the B11-Fc mAb at a dose of 0.6 mg/kg provided 100% protection from poisoning with BoNT/A that was simultaneously administered at a dose of 20 LD50. The study determined the main pharmacokinetic parameters of the B11-Fc mAb. The experiments demonstrated that prophylactic administration of the B11-Fc mAb for 21 days had a protective effect against BoNT/A administered intraperitoneally at a dose of 5 LD50, and therapeutic administration of the mAb 14 h after intragastric administration of the toxin at a dose of 12,000 intraperitoneal LD50 provided 100% protection.
CONCLUSIONS. The authors optimised the technology for laboratory-scale production of the candidate modified single-domain mAb. In vivo experiments conducted using BoNT/A toxicity models demonstrated that the B11-Fc mAb is highly effective in botulism prevention and treatment. On the basis of preclinical data, phase I clinical trials have been initiated to study B11-Fc in healthy volunteers.
STANDARDIZATION AND QUALITY CONTROL

INTRODUCTION. The advancement of manufacturing technologies and the expanding range of biological medicinal products (BMPs) and indications for their use necessitate the development of a unified approach to BMP standardisation at both national and regional levels. In the context
of the common pharmaceutical market of the EAEU (EAEU), the requirements for medicinal products should take into account the national pharmacopoeial quality standards, which, in turn, are subject to harmonisation with regional pharmacopoeial standards.
AIM. This study aimed to systematise the requirements for BMPs of the State Pharmacopoeia of the Russian Federation and the Pharmacopoeia of the EAEU to achieve harmonisation of the national and regional standards.
DISCUSSION. This study analysed special considerations for BMP standardisation in accordance with the national pharmacopoeias of the EAEU Member States. In addition, the study covered the requirements of major international pharmacopoeias and regulatory documents, including those by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Having established the basic principles for formulating the requirements for BMPs within the scope of the Pharmacopoeia of the EAEU, the authors drafted a general chapter for BMPs for the Pharmacopoeia of the EAEU. This draft general chapter introduced a uniform definition of BMPs, taking into account regional and national legislation. The general chapter standardised compendial requirements for different groups of BMPs, highlighting the main considerations for the production (technology) and quality control of intermediates and finished medicinal products. When harmonising the State Pharmacopoeia of the Russian Federation with the requirements of the Pharmacopoeia of the EAEU, the authors outlined the main approaches to BMP standardisation applied by the State Pharmacopoeia of the Russian Federation and made amendments to General Chapter 1.7.1.0010.18 Biological medicinal products of the national pharmacopoeia. In particular, the authors removed the requirements for BMP production that were not universally applicable to all BMPs, along with the lists of mandatory tests for active pharmaceutical substances and finished medicinal products.
CONCLUSIONS. The study systematised the requirements for BMPs outlined in international and regional regulatory documents. The authors drafted the general chapter for BMPs for the Pharmacopoeia of the EAEU and used it in updating General Chapter 1.7.1.0010.18 Biological medicinal products of the State Pharmacopoeia of the Russian Federation. The study also substantiated the approaches to drafting compendial texts (general chapters and monographs) for BMPs.

INTRODUCTION. Currently, diphtheria, pertussis, and tetanus (DTP) vaccines are available in tetra-, penta-, and hexavalent combinations with inactivated poliomyelitis, Haemophilus influenzae, and hepatitis B components. Despite the widespread introduction of DTP vaccines in national vaccination programmes, concerns remain about the immunogenicity and safety of the pertussis component, the standardisation of vaccine production and quality control methods, and the inclusion of DTP vaccines in national routine vaccination schedules.
AIM. This study aimed to provide an updated overview of DTP-based combined vaccines and analyse the current challenges associated with their use and quality improvement.
DISCUSSION. DTP vaccines hold a central place in national routine vaccination schedules. The development of numerous safe and effective DTP vaccines has contributed to the formulation of DTP-based combined vaccines that include additional components and are suitable for infants. The addition of inactivated components against poliomyelitis, H. influenzae, and hepatitis B has facilitated the introduction of DTP-based combined vaccines into the recommended vaccination programmes and has reduced the number of injections received by a child. However, DTP-based combined vaccines from different manufacturers differ in the composition and quantity of antigens and in quality control methods. The key differences in the composition of these vaccines are due to the inclusion of either whole-cell or acellular pertussis. The current global rise in the incidence of pertussis is associated with the widespread use of acellular vaccines, which do not induce long-term immunity. This review considers the mutual influence of antigens in relation to vaccine efficacy and safety and addresses the standardisation issues associated with antigen production and quality control. The article analyses data on various DTP-based combined vaccines and the quantity of antigens in them. The review discusses promising areas for further improvement of the quality and effectiveness of DTP-based combined vaccines.
CONCLUSIONS. Addressing unresolved standardisation issues in the production and quality control of DTP-based combined vaccines, which (along with country-specific licensing requirements) limit the international exchange of vaccines, can facilitate international recognition and ensure a high level of potency and safety of DTP-based combined vaccines.

INTRODUCTION. High quality standards for sterilised water for injections arise from the need to guarantee the safety and effectiveness of injectable medicines, especially biologicals, since the presence of impurities in the solvent (mainly microbial contaminants, endotoxins, and heavy metals) can lead to serious adverse drug reactions. Therefore, it is important to determine the most promising approaches to assessing the quality of water for injections to develop the regulatory requirements for the Eurasian Economic Union (EAEU).
AIM. This study aimed to analyse key trends in the quality assessment of water for injections used as a solvent for medicinal products.
DISCUSSION. This study involved a retrospective comparison of the quality control requirements for water for injections established by the world’s major pharmacopoeias, including the State Pharmacopoeia of the Russian Federation, the United States Pharmacopeia, the Japanese Pharmacopoeia, the European Pharmacopoeia, the British Pharmacopoeia, the Pharmacopoeia, and the Pharmacopoeia of the People’s Republic of China. Additionally, the comparison included recommendations by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use and the Pharmacopoeia Discussion Group. The past decade witnessed significant changes in the approach of international pharmacopoeias to the control of organic and inorganic impurities in water for injections, both in terms of analytical procedures and in terms of the number of tests required. According to the comparison, the most significant changes to the quality control requirements for sterilised water for injections in the finished dosage form were introduced by the European Pharmacopoeia in 2024. The authors considered the possibility to streamline the quality control procedure by reducing the number of tests and replacing the currently required ten qualitative tests for inorganic impurities with a single quantitative determination of electrical conductivity. This article describes the replacement of the test for organic impurities with a quantitative test for total organic carbon. Furthermore, this article presents the quality control results obtained at the Scientific Centre for Expert Evaluation of Medicinal Products of the Ministry of Health of the Russian Federation for 148 batches of sterilised water for injections supplied with biologicals produced by 38 Russian and international manufacturers.
CONCLUSIONS. The current requirements of the revised monograph for water for injections and sterilised water for injections of the European Pharmacopoeia (07/2024:0169) may inform drafting a relevant compendial standard for the EAEU and updating the monograph for water for injections of the State Pharmacopoeia of the Russian Federation. This will help optimise the quality control procedures, increase the speed and accuracy of testing, and reduce financial and labour costs, which will improve the quality of the solvent and the associated medicinal products. The adoption of these requirements will contribute to pharmacopoeial harmonisation by setting uniform quality control criteria for water for injections for both national and international manufacturers, which will reduce regulatory barriers and facilitate the entry of the solvent dosage form into international markets.
PRECLINICAL STUDIES
INTRODUCTION. Although whole-cell pertussis vaccine (WCPV) is highly effective, its widespread use is limited by adverse reactions (increased body temperature, administration site oedema, allergic reactions, and febrile seizures). Therefore, there is a need to explore strategies for reducing the toxicity of WCPV.
AIM. This study aimed to evaluate the toxicity of WCPV in mice after storing WCPV under different conditions.
MATERIALS AND METHODS. This study focused on Bordetella pertussis strains, including vaccine production strains and circulating strains isolated from children with pertussis. Ten WCPV samples were prepared by washing the grown cultures and adding formaldehyde (inactivating agent) and thimerosal (preservative) to the bacterial suspensions. Outbred mice were randomised into 10 test groups and 1 control group (10 animals per group). The test groups received intraperitoneal injections of the WCPV samples, while the control group received 0.9% sodium chloride with thimerosal. Mice were weighed before injection, as well as on Days 1 and 7 after injection. The weighing was timed to coincide with the peak activity of the main B. pertussis toxins, lipo-oligosaccharide (Day 1) and pertussis toxin (Day 7). The specific safety index was calculated as the ratio of the weight gain of the test animals to the weight gain of the control animals (in %) and was monitored for the WCPV samples throughout the WCPV shelf life (12 months).
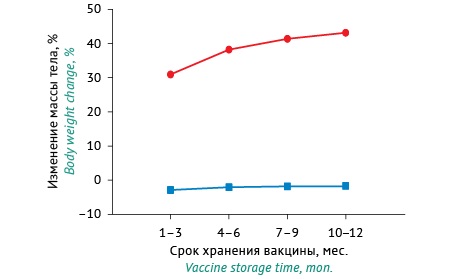
RESULTS. On Day 1 after injection, the B. pertussis suspensions kept with the inactivating agent under recommended storage conditions for 1–3 months caused a 2.84% decrease in the body weight of mice from the baseline weight, and the suspensions inactivated for 10–12 months caused a 1.62% weight loss relative to the baseline. On Day 7 after injection, the mice that received the B. pertussis suspensions inactivated for 1–3 months showed a 31.0% increase from the baseline weight, and the mice that received the suspensions inactivated for 10–12 months demonstrated a 43.22% weight gain. The suspensions inactivated for 1–3 months and 10–12 months had specific safety indices of 69.38% and 84.31%, respectively. According to these findings, the residual toxicity of WCPV decreased after 12 months of storage, and the process of pertussis toxin inactivation lasted throughout the entire WCPV shelf life. Spearman’s correlation coefficient, characterising the strength of the relationship between the specific safety index and the weight gain of mice, was 0.55 (p<0.01) on Day 7 (a noticeably strong relationship) and 0.349 (p<0.01) on Day 1 (a moderately strong relationship).
CONCLUSIONS. The results suggest a more complete detoxification of pertussis toxin in the samples of WCPV kept with the inactivating agent under recommended storage conditions for at least 10–12 months compared with that after a shorter period. To improve the safety of DTP vaccines, it is advisable to use WCPV batches stored for 10–12 months.

ISSN 2619-1156 (Online)































