



ISSUE TOPIC: INNOVATIVE IMMUNOBIOLOGICALS
INTRODUCTION. The current progressive increase in pertussis incidence and infant mortality rates is due to the insufficient effectiveness of existing vaccines, both in Russia and worldwide. Previous clinical trials showed that healthy adult volunteers developed long-term antibacterial immunity after vaccination with GamLPV, an intranasal recombinant live pertussis vaccine developed by the National Research Center for Epidemiology and Microbiology named after the honorary academician N. F. Gamaleya. Further clinical development of GamLPV in paediatric volunteers, including infants, requires preclinical studies in a newborn monkey model.
AIM. This study aimed to evaluate the safety, immunogenicity, and protective efficacy of the GamLPV vaccine in infant hamadryas baboons (Papio hamadryas) challenged with pertussis after intranasal vaccination.
MATERIALS AND METHODS. The study used 20 hamadryas baboons, including 7 infants aged 1–1.5 months, 7 mothers of these infants, and 6 control animals. The study examined the time course of changes in serum levels of specific IgG antibodies to pertussis toxin (PT) and filamentous haemagglutinin (FHA) by enzyme-linked immunosorbent assay (ELISA), monitored changes in serum levels of Bordetella pertussis antibodies by agglutination immunoassay, and detected B. pertussis DNA in oropharyngeal aspirates by real-time polymerase chain reaction.
RESULTS. Intranasal GamLPV administration to infant baboons induced the production of specific IgG antibodies to PT and FHA (ELISA) and an increase in the total pertussis antibody titre (agglutination immunoassay). GamLPV did not cause any injection site or systemic reactions. There were no changes in complete blood counts and serum biochemistry profiles after vaccination. The protective efficacy of GamLPV against B. pertussis was demonstrated in challenge tests, where immunised animals had no clinical signs or laboratory findings indicative of pertussis in contrast to controls.
CONCLUSIONS. The study demonstrated the safety and immunogenicity of the intranasal live pertussis vaccine GamLPV in newborn hamadryas baboons. GamLPV shows promise in the primary vaccination of infants, the revaccination of children and adults, and the development of herd immunity against pertussis in families.
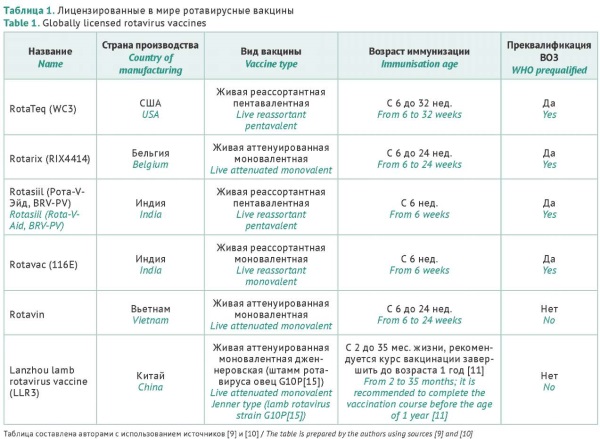
INTRODUCTION. Vaccination is recognised as the only effective method for preventing rotavirus disease. Rotavirus remains a leading cause of death in young children, mainly, in developing countries. Currently, oral rotavirus vaccines for infant immunisation are available worldwide, and novel types of rotavirus vaccines are also under development, in particular, in the Russian Federation. However, there are no regulations or guidelines helping developers to design an optimal preclinical and clinical programme for rotavirus vaccines.
AIM. This study aimed to analyse and summarise global experience in planning and conducting preclinical and clinical studies of rotavirus vaccines in order to provide recommendations for national vaccine developers.
DISCUSSION. This study presents an analysis of the available data (and, specifically, the data obtained for the past five years) on all rotavirus vaccines used in the world that have been clinically proven to be effective in preventing severe rotavirus gastroenteritis and reducing the number of hospital admissions due to acute intestinal infections. The effectiveness of rotavirus vaccines varies in different regions of the world and may be lower in developing countries for various reasons. The safety profile of oral rotavirus vaccines is generally considered favourable. Nevertheless, there are still some concerns regarding intestinal intussusception in infants following vaccination. To address the abovementioned problems, researchers, including those in Russia, are developing novel types of rotavirus vaccines, predominantly focusing on inactivated (subunit or recombinant) preparations. For planning and conducting preclinical studies of a rotavirus vaccine, it is advisable to adopt general approaches that involve assessing the acute and chronic toxicity, immunogenicity, and safety pharmacology of the rotavirus vaccine and the virus-neutralising activity of vaccination-induced antibodies. Clinical trials of a rotavirus vaccine should assess its effectiveness in preventing rotavirus gastroenteritis of any severity, hospitalisation, and acute viral intestinal infections of any aetiology in the target age group of young children. Furthermore, it is important to confirm the safety of the rotavirus vaccine and demonstrate the absence of mutual interference with the immunogenicity of the rotavirus vaccine and other vaccines co-administered in the vaccination schedule.
CONCLUSIONS. Preclinical studies of rotavirus vaccines may use standard and generally accepted approaches. However, planning and conducting clinical trials requires specific considerations associated with both the nature of rotavirus infection and the national infant vaccination schedule.
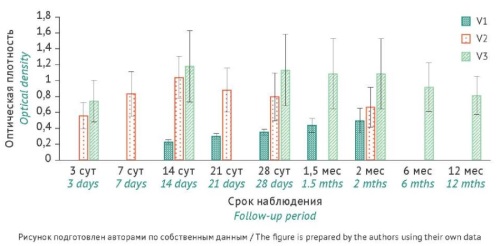
INTRODUCTION. Rotavirus vaccines based on virus-like particles (VLPs), non-infectious recombinant proteins of human rotavirus A that mimic the structure of the native virus, show promise for preventive vaccination. Presumably, the optimal method to determine the potency of VLP-based rotavirus vaccines is the enzyme-linked immunosorbent assay (ELISA) method used to measure the titre of specific IgG antibodies to rotavirus A proteins in serum samples from vaccinated animals.
AIM. This study aimed at assessing the potency of a VLP-based rotavirus vaccine by developing and validating an analytical procedure using ELISA to determine the levels of antibodies to the VP2 and VP6 proteins of rotavirus A in serum samples from vaccinated guinea pigs.
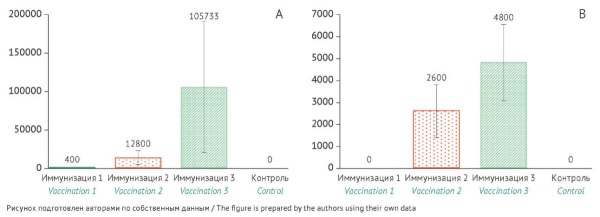
MATERIALS AND METHODS. The potency of the VLP-based rotavirus vaccine was determined in vivo in three types of experimental animals, including BALB/c mice, agouti guinea pigs, and newborn minipigs. The animals received three intramuscular injections of the vaccine at a dose of 30 µg. This study used the indirect ELISA method to quantify VP2- and VP6-specific immunoglobulin G (IgG) antibodies and the virus-neutralisation test to measure neutralising antibodies (nAbs) to rotavirus A in animal serum samples. The study involved calculating the geometric mean titres (GMTs) of antibodies. The authors validated the analytical procedure for potency assessment on two batches of the vaccine using standard statistical analysis methods.
RESULTS. The study compared VP2- and VP6-specific IgG and nAb levels 14 days after the first, second, and third vaccinations. The authors observed a significant increase in antibody titres and statistically significant (p<0.05) differences between groups as early as after the second vaccination. Double vaccination induced rotavirus-specific IgGs and nAbs in newborn minipigs (GMTs of 200.0 and 108.9, respectively) and guinea pigs (GMTs of 12,800 and 2,600, respectively). Vaccinated mice demonstrated a significant increase in rotavirus-specific IgG levels (GMT of 572,440). Guinea pigs were selected as a relevant model for validating the ELISA-based potency assessment procedure. The validation study used a double vaccination scheme. The validation using two batches of the VLP-based rotavirus vaccine indicated that the ELISA-based analytical procedure met the acceptance criteria for specificity, repeatability, and intermediate precision. The repeatability assessment resulted in a coefficient of variation (CV) of 12.4% for batch 1 and a CV of 7.7% for batch 2, whereas the intermediate precision assessment showed a CV of 6.9% for batch 1 and a CV of 10.2% for batch 2, which were within the acceptance criteria for both validation parameters (CV≤15%).
CONCLUSIONS. The authors developed and validated an ELISA-based analytical procedure for assessing the potency of VLP-based preventive rotavirus vaccines. According to the study results, ELISA is applicable to the control of the potency of VLP-based preventive rotavirus vaccines.
INTRODUCTION. Currently, rotavirus infection is prevented with live attenuated vaccines. However, international and Russian vaccination practices, as well as the physiological characteristics of paediatric patients, necessitate the development of inactivated rotavirus vaccines. Prerequisites for the development of such vaccines are the availability of virus strains capable of stable replication and the selection of optimal inactivation conditions providing for the required antigenicity and immunogenicity levels.
AIM. This study aimed to evaluate and compare the characteristics of the rotavirus-specific immune response to native strains and to a composition of inactivated rotavirus A strains in a mouse model.
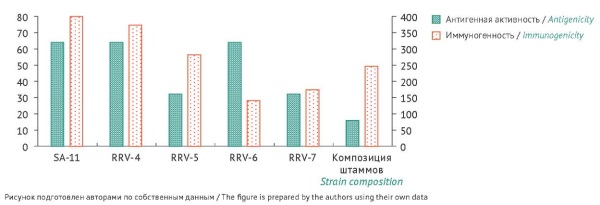
MATERIALS AND METHODS. The study used human rotavirus A strains (RRV-4, RRV-5, RRV-6, and RRV-7), a standard rotavirus strain (SA-11 NVC 2364, National Virus Collection of the Russian Federation), and cultures of pig embryo kidney cells treated with Versene solution (SPEV) and Vero cells. Virus titration was used to determine the infectivity of the strains grown in Vero cells maintained in continuous culture. The authors monitored infected cell cultures up to the onset of the cytopathic effect, calculated the 50% tissue culture infectious dose (TCID50) by the Kärber method modified by Ashmarin, and expressed the results as log10 TCID50/mL. Virus strains were inactivated with formaldehyde. To evaluate immunogenicity, outbred white mice were immunised with native strains and the composition of inactivated strains (RRV-4, RRV-5, RRV-6, and RRV-7). After immunisation, blood was taken from the animals, and the serum titre of rotavirus A antibodies was determined by indirect heterogeneous enzyme immunoassay.
RESULTS. The infectivity of the rotavirus strains adapted to Vero cells ranged from 8.9 to 7.9 log10 TCID50/mL. When selecting inactivation conditions, the authors showed that inactivation occurred at a temperature of 37 °C and a formaldehyde concentration of 0.05–0.025% (depending on the duration of treatment). The antigenicity analysis demonstrated that the antigen titre of the inactivated strain composition (1:16) was lower than that of native strains (1:32–1:64). The authors demonstrated comparability of immunogenicity profiles of the inactivated strain composition and native strains in mice.
CONCLUSIONS. The study generated candidate rotavirus A strains that exhibited stable replication in continuous cultures of Vero cells. The authors selected optimal inactivation conditions for these rotavirus strains and developed an inactivated strain composition showing antigenicity and immunogenicity. The presented data suggest that the composition of inactivated rotavirus A strains can be considered as a basis for further development of an inactivated rotavirus vaccine.
ANTIVIRAL MEDICINAL PRODUCTS
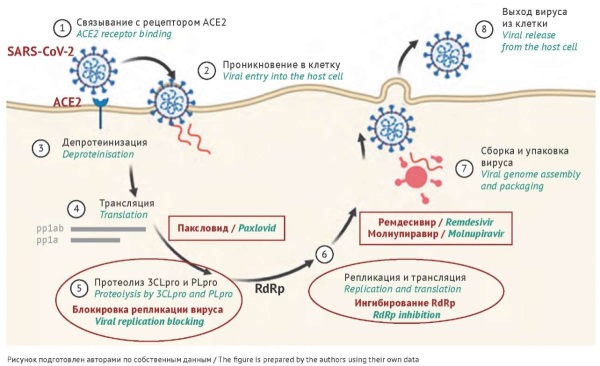
INTRODUCTION. The high prevalence of mutations in the SARS-CoV-2 genome raises particular concerns about the resistance of the virus to current antiviral therapy, including inhibitors of the main protease, or 3C-like protease (3CLpro), and RNA-dependent RNA polymerase (RdRp).
AIM. This study aimed to analyse the prevalence, spectrum, and causes of SARS-CoV-2 mutations conferring resistance to approved and pipeline RdRp and 3CLpro inhibitors on the basis of clinical, virological, and genotypic data.
DISCUSSION. The authors have analysed the prevalence of SARS-CoV-2 mutations conferring resistance to antivirals (RdRp inhibitors, including remdesivir and molnupiravir, and 3CLpro inhibitors, including paxlovid) in 2021–2024. The results suggest that certain mutations existed prior to the use of these antivirals. The prevalence of resistance-conferring mutations does not exceed 0.5% of the global population. However, the results of clinical and experimental studies demonstrate the possibility of a more than 200-fold reduction in susceptibility to medicinal products and, in particular, the emergence of multidrug-resistant variants. This is especially important for immunocompromised patients. SARS-CoV-2 can persist in such patients for many months, during which spontaneous or selection-driven mutations can render antiviral therapy ineffective. This would create a risk of spreading drug-resistant variants and/or a risk of adverse outcomes for patients.
CONCLUSIONS. As COVID-19 treatment coverage increases, there may be a rise in drug-resistant variants of the virus. The presented data indicate the need for genomic epidemiological surveillance, including an analysis of potential targets for medicinal products based on clinical observations. In the future, surveillance data may determine the treatment strategy and the need to develop new antivirals (RdRp and protease inhibitors) adjusted to resistant SARS-CoV-2 variants.
CELL THERAPY
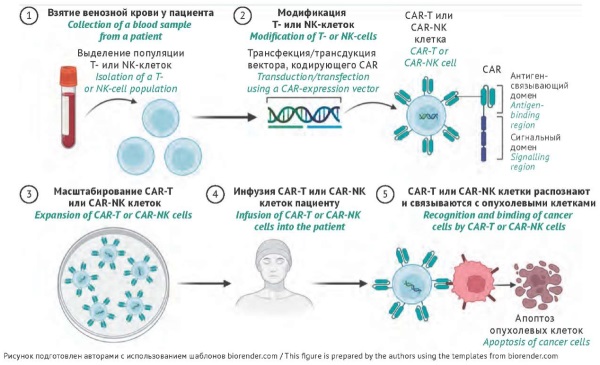
INTRODUCTION. Cell therapies and tissue-engineered products are aimed at patients with severe conditions (genetic and neurodegenerative disorders, cancers, musculoskeletal injuries, burns, etc.) that lack alternative treatment options. Analysis of clinical efficacy data on cell-based medicinal products is important for understanding their translational potential in personalised medicine.
AIM. This study aimed to review key trends in cell therapy, analyse data on approved cell therapies and tissue-engineered products, and assess challenges and prospects for their use.
DISCUSSION. This article analyses data on the composition of cell therapies and tissue-engineered products, indications for their use, and the results of clinical studies. Cell-based medicinal products are derived from autologous or allogeneic mesenchymal and limbal stem cells, epithelial cells, chondrocytes, native or genetically engineered haematopoietic stem cells, genetically engineered lymphocytes (CAR-T, CAR-NK), etc. Medicinal products based on cell technologies have been approved in many countries, including the USA (approximately 30), the European Union (approximately 20), Japan (18), South Korea (15), etc. As of today, two cell therapies have been granted marketing authorisation in the Russian Federation. The first is based on CAR-T cells (a gene therapy product), and the other is based on chondrocytes (a cell-based medicinal product); the latter has been developed in Russia. The main advantages of cell therapy products include higher efficacy and fewer adverse drug reactions in comparison with standard treatment modalities. The main challenges of cell therapy include the risks of immune reactions and mutagenesis associated with lentiviral vectors or CRISPR/Cas9 technology, as well as limited efficacy of CAR-T and CAR-NK cells due to immunosuppressive properties of tumour microenvironment.
CONCLUSION. In comparison with conventional treatment approaches, the use of cell therapies and tissue-engineered products can help effectively eliminate defects in various body tissues, avoid highly invasive surgical interventions, and reduce regeneration time. Thus, ensuring development of similar but at the same time more affordable Russian medicinal products can bring great benefits for the healthcare system of the Russian Federation.
QUALITY CONTROL
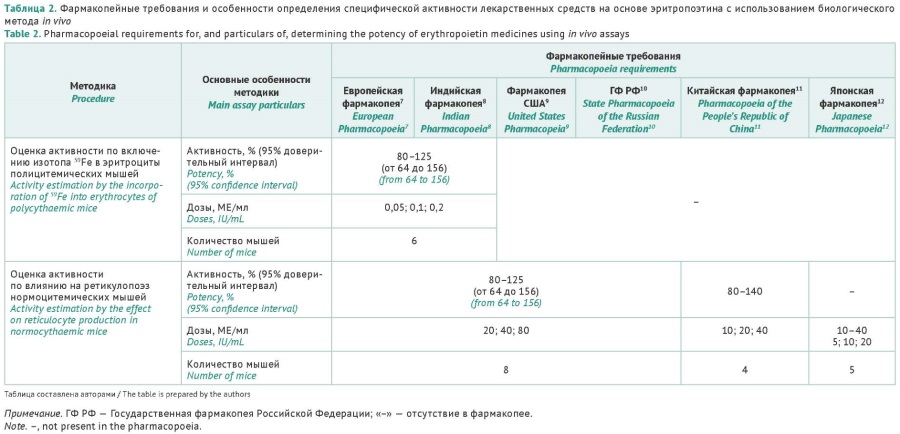
INTRODUCTION. Given the widespread clinical use of recombinant human erythropoietin (rhEPO) products from different manufacturers, potency assays should ensure that patients receive comparable doses of rhEPO across medicinal products. The harmonisation of approaches to potency testing requires the use of pharmacopoeial bioassays and appropriate international/pharmacopoeial reference standards (RSs).
AIM. This study aimed to summarise information on pharmacopoeial requirements, relevant RSs, and bioassays (in vivo and in vitro) for the assessment of rhEPO potency, as well as to analyse the pharmacopoeial compliance of manufacturers’ specifications for rhEPO products authorised in Russia.
DISCUSSION. This article presents information on the molecular structure of erythropoietin. The glycosylation profile of erythropoietin not only accounts for most of the differences in the half-life and biodegradation rate but also significantly influences the potency of rhEPO products. The authors outlined the pharmacopoeial requirements for potency assays in vivo, summarised the information on RSs for potency determination, characterised the development of potency assays in vitro, and studied the possibility of including in vitro assays in pharmacopoeias. The analysis showed that some potency assays used for rhEPO products manufactured in Russia did not comply with the requirements of the State Pharmacopoeia of the Russian Federation.
CONCLUSIONS. The study identified the need to develop and certify a national pharmacopoeial RS for the potency of rhEPO in order to satisfy the demands of Russian manufacturers in the context of import substitution. To implement adequate 3R-compliant methods for rhEPO quality assessment, it is necessary to harmonise approaches to potency testing of rhEPO products and develop a consolidated document governing such testing.
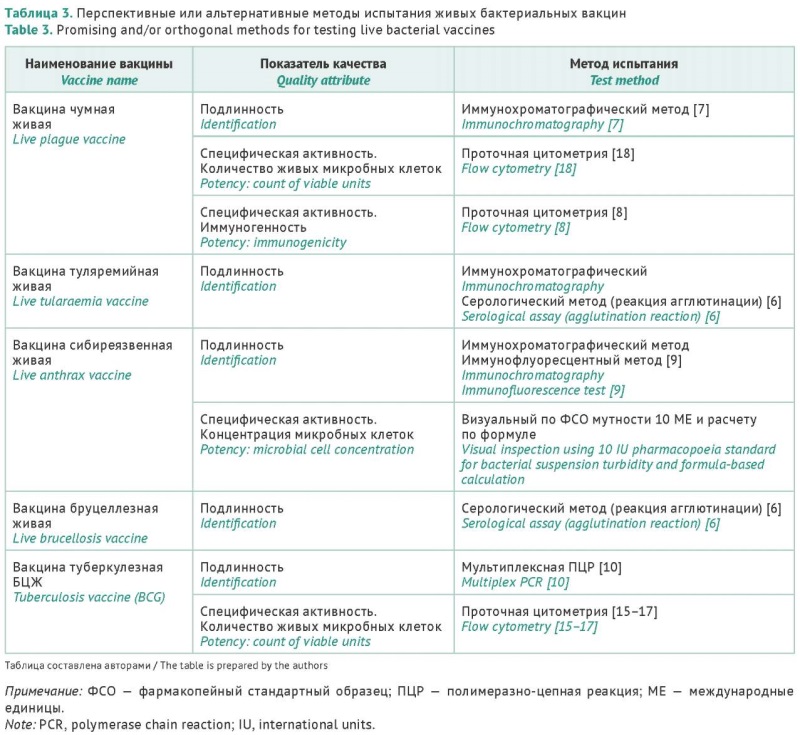
INTRODUCTION. Currently, there are live bacterial vaccines (LBVs) for the prevention of tuberculosis and particularly dangerous infections, such as plague, tularaemia, anthrax, and brucellosis. LBVs provide reliable and long-lasting protection. The high effectiveness of LBVs is due to their ability to mimic natural infections. Despite significant differences in LBV compositions, indications, and production species, the applicable quality control approaches share some common principles. At the same time, there are significant differences in methodological approaches to the assessment of a number of quality attributes, and some of these approaches require improvement and/or optimisation.
AIM. This study aimed to analyse current regulatory requirements for the quality of LBVs having different compositions and indications, to identify the quality attributes of LBVs with the associated quality control methods in need of improvement, and to explore promising pathways for further development in this area.
DISCUSSION. The review of regulatory standards and manufacturer specifications identified several quality attributes common to all LBVs. Vaccine identity was considered a key quality attribute. Motivated by the development of novel national diagnostic test systems, the authors analysed the performance of these test systems in practice to determine their potential for the identification of bacterial vaccines. In particular, immunochromatographic and serological techniques were successfully applied to the quality control of the tularaemia vaccine, a serological method was deemed suitable for brucellosis vaccines, and immunochromatography was recommended for plague and anthrax vaccines. A multiplex PCR method passed practical testing to be used for the quality control of BCG vaccines with reagents from Russian manufacturers. Flow cytometry was considered a promising technique for improving the quality control of LBVs in terms of viable units (tuberculosis and plague vaccines) and immunogenicity (plague vaccines).
CONCLUSIONS. This analysis of regulatory requirements highlighted problematic issues related to the standardised quality attributes of LBVs and methods for their assessment. The literature review results and the authors’ own data indicate promising directions for improving the methodology of LBV quality assessment. One of these promising directions is the use of Russian-made commercial test systems for LBV identification.
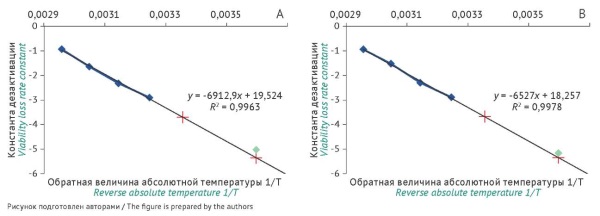
INTRODUCTION. A key priority in maintaining a collection of microorganisms is to ensure the stability of characteristics and the viability of microbial cultures during their storage and transport. In addition, some applications of collection strains as control samples require accurate data on the number of viable microbial cells in each sample. Therefore, it is necessary to develop and implement an analytical procedure for predicting the guaranteed shelf life of test strains.
AIM. This study aimed to predict the guaranteed shelf life for test strains in a variety of primary packaging by assessing changes in microbial viability under accelerated storage conditions, with the Salmonella enterica subsp. enterica serovar Abony NCTC 6017 strain as a model organism.
MATERIALS AND METHODS. The study used lyophilised samples of the S. enterica subsp. enterica serovar Abony NCTC 6017 strain deposited in the National Collection of Pathogenic Microorganisms (NCPM) at the Scientific Centre for Expert Evaluation of Medicinal Products of the Ministry of Health of the Russian Federation. The studied primary packaging types included vacuum-filled borosilicate glass ampoules and 2R lyophilisation vials. The samples were tested for a number of quality attributes (loss on drying, viable cell count, cell viability, colony morphology, and biochemical identification) and subjected to accelerated shelf-life testing at elevated temperatures (35–65 °С).
RESULTS. The study did not show any significant differences in the quality of lyophilised samples depending on the type of primary packaging. The authors experimentally determined rate constants for the loss of viability in microbial cultures during storage at elevated temperatures and calculated the rate constants for the storage and transport temperatures and for different types of primary packaging. The predicted time to viable cell count reduction to 10% of the initial level was 19 years for vials and 25 years for ampoules, and the predicted time to 50% viability was 5.8 years for vials and 7.6 years for ampoules.
CONCLUSIONS. The results of this study confirm the applicability of different primary packaging (ampoules and vials) for the lyophilisation and storage of microbial test strains. The data obtained can guide further research and contribute to the development of recommendations for the storage of lyophilised strains in various types of packaging.

ISSN 2619-1156 (Online)































