



ОСНОВНАЯ ИНФОРМАЦИЯ О ЖУРНАЛЕ
«БИОпрепараты. Профилактика, диагностика, лечение» – научно-практический рецензируемый журнал открытого доступа, выпускаемый в печатной и онлайн-версиях, единственный в России, посвященный полному циклу разработки биологических лекарственных препаратов.
Цель журнала: освещение актуальных вопросов разработки регуляторных процедур, стандартизации, контроля качества, производства терапевтических, профилактических и диагностических биологических лекарственных препаратов, биомедицинских клеточных продуктов и их применения, включая клинические аспекты, для профилактики, диагностики и лечения инфекционных заболеваний, изучения аллергических и иммунопатологических процессов.
Более подробная информация – в разделе «Цели и тематика».
Целевая аудитория: биотехнологи, иммунологи, вирусологи, микробиологи, специалисты в области молекулярной и клеточной биологии, фармакологи, представители экспертных и регуляторных учреждений, специалисты в области биофармацевтической индустрии.
Учредитель и издатель: ФГБУ «Научный центр экспертизы средств медицинского применения» Министерства здравоохранения Российской Федерации.
Периодичность: 4 раза в год.
Импакт-фактор: двухлетний импакт-фактор РИНЦ (2023) – 0,860.
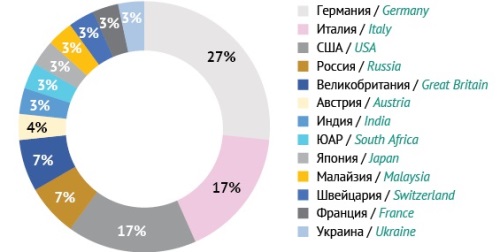
Редколлегия. Географическое разнообразие:
- 2 континента
- 7 стран
- 12 городов
Рецензирование:
- Двойное слепое
- Минимум 2 рецензента на рукопись
Основные метрики журнала:
- 8 дней в среднем от подачи до первого решения
- 177 день в среднем от подачи до онлайн публикации
- 31% приглашенных авторов
- 74% доля принятия рукописей
- 25 тыс. загрузок PDF в 2023 г.
Плата за публикацию: бесплатно.
Индексация. Входит в Scopus (Accepted Titles May 2025), РИНЦ, Перечень ВАК, Russian Science Citation Index, DOAJ. Информация об индексации в других российских и международных базах доступна в разделе Индексирование.
Регистрация. Свидетельство о регистрации средства массовой информации ПИ № ФС77-82918 от 14.03.2022 г.
Подписка. Подписной индекс в каталоге Пресса России – 57941, Урал-Пресс – 57941.
Текущий выпуск

ТЕМА НОМЕРА: ТРЕНДЫ КОНТРОЛЯ КАЧЕСТВА И СТАНДАРТИЗАЦИИ БИОЛОГИЧЕСКИХ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ

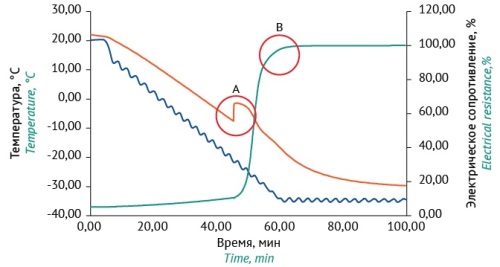
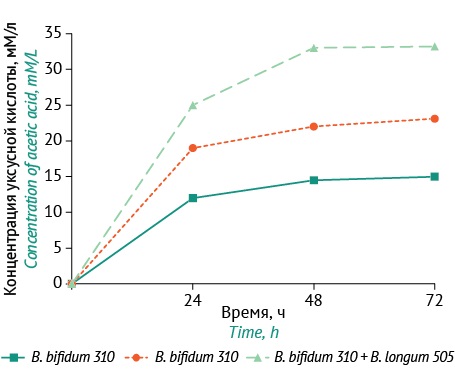
ПРОБИОТИКИ
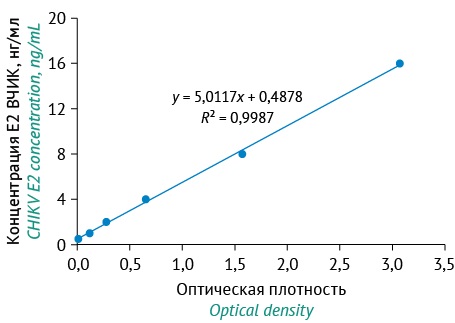
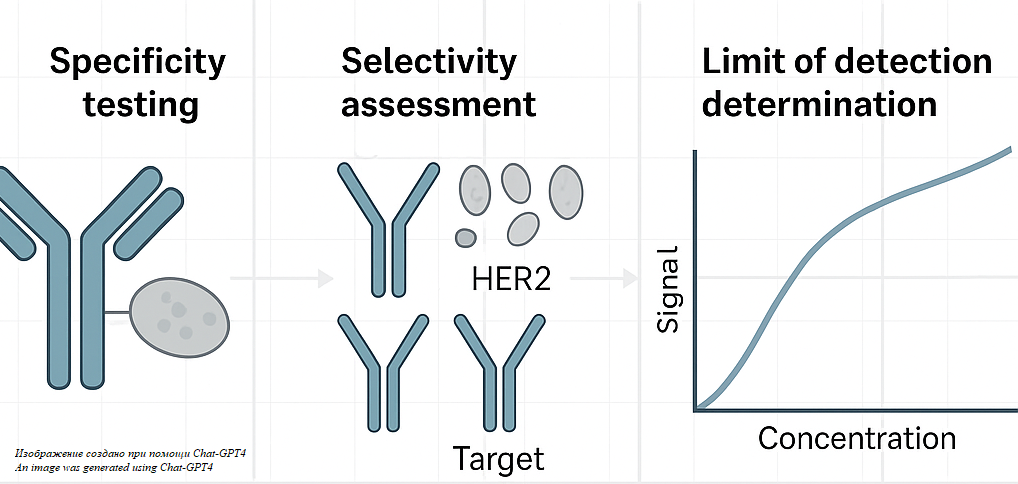
РАЗРАБОТКА И ВАЛИДАЦИЯ МЕТОДИК
Новости
2025-06-03
Поздравляем с избранием в академики РАН!
Редколлегия и редакция журнала «БИОпрепараты. Профилактика, диагностика, лечение» поздравляют члена редколлегии – Мусу Рахимовича Хаитова, избранного в действительные члены Российской академии наук.
| Еще объявления... |



























