




Содержание
Перейти к:
https://doi.org/10.30895/2221-996X-2024-24-1-46-60
Перейти к:
АКТУАЛЬНОСТЬ. При контроле качества протеолитических ферментов, входящих в состав лекарственных препаратов, активность протеаз определяется спектрофотометрически, измерением амидазной или эстеразной активности с синтетическим субстратом, и протеолитической по методу Ансона. Данные методы требуют специальных субстратов, обладают низкой чувствительностью, а их специфичность может оказаться недостаточной и приводить к серьезным ошибкам. Альтернативный подход определения активности протеаз реализуется путем количественного масс-спектрометрического измерения, которое обеспечивается добавлением к продуктам гидролиза испытуемого фермента пептида с изотопной меткой. Такой подход позволяет определять протеолитическую активность, причем по гидролизу конкретных пептидных связей, одновременно с подтверждением подлинности и специфичности испытуемого образца с высокой чувствительностью и без использования специальных субстратов.
ЦЕЛЬ. На примере трипсина и казеина исследовать возможность определения активности фермента методом количественной масс-спектрометрии с меткой ¹⁸О одновременно с подтверждением подлинности.
МАТЕРИАЛЫ И МЕТОДЫ. Использовали трипсин, казеин, H₂¹⁸O (НПО «Изотоп», Россия). Разделение пептидов проводили с помощью системы ВЭЖХ Agilent 1100, масс-спектры получали с использованием масс-спектрометра MALDI-TOF/TOF Bruker Ultraflex II. Количественный масс-спектрометрический анализ проводили, применяя пептид сравнения. Пептид получали путем гидролиза казеина трипсином с последующей очисткой с помощью ВЭЖХ. Для получения пептида с изотопной меткой ¹⁸О исходный пептид высушивали и инкубировали в воде H₂¹⁸О. Количественное определение продукта проводили с применением масс-спектрометрии MALDI-TOF. Для определения активности фермента и расчета константы Михаэлиса (КМ) использовали метод количественной масс-спектрометрии с меткой ¹⁸О.
РЕЗУЛЬТАТЫ. При гидролизе казеина трипсином идентифицированы фрагменты, соответствующие цепям этого белка. Получен изотопно-меченый стандарт, концентрация которого была рассчитана масс-спектрометрически. Определена скорость гидролиза казеина трипсином и рассчитана КМ для трипсина, которая составила 13,65±0,60 мкМ. Стандартное отклонение значений повторных измерений показало, что ошибка измерений оказалась меньше по сравнению со спектрофотометрическим методом, а минимальная установленная чувствительность предложенного метода составила 0,50±0,08 мкМ.
ВЫВОДЫ. Подтверждена возможность применения метода количественного масс-спектрометрического анализа с меткой ¹⁸О для определения активности ферментов на примере трипсина. Установленная чувствительность позволяет оценивать активность фермента одновременно с определением его подлинности и специфичности. Предложенный подход является универсальным, не требует дорогостоящих материалов и реактивов, а также может быть легко адаптирован для определения активности практически любой протеазы.
Константинов М.А., Жданов Д.Д., Торопыгин И.Ю. Количественная масс-спектрометрия с меткой ¹⁸О как альтернативный подход к определению активности протеаз на примере трипсина. БИОпрепараты. Профилактика, диагностика, лечение. 2024;24(1):46-60. https://doi.org/10.30895/2221-996X-2024-24-1-46-60
Konstantinov M.A., Zhdanov D.D., Toropygin I.Yu. Quantitative mass spectrometry with ¹⁸O labelling as an alternative approach for determining protease activity: an example of trypsin. Biological Products. Prevention, Diagnosis, Treatment. 2024;24(1):46-60. (In Russ.) https://doi.org/10.30895/2221-996X-2024-24-1-46-60
К настоящему времени сформировалась достаточно широкая номенклатура лекарственных препаратов белковой природы, полученных как из природных источников (органы и ткани человека и животных), так и биотехнологическими методами (рекомбинантные белки и их производные). В их числе моноклональные антитела, гормоны, цитокины, интерлейкины, интерфероны (ИФН-α, ИФН-β, ИФН-γ), ингибиторы факторов роста, гибридные белки и др. [1][2].
Такие препараты являются, вероятно, самыми сложными для аналитического контроля. Согласно требованиям Государственной фармакопеи Российской Федерации (ГФ РФ)1 — ОФС.1.7.2.0035.18, ОФС.1.7.1.0007.15, ОФС.1.7.1.0014.18 — допускаются различные методы подтверждения подлинности и активности: от прямой проверки специфичности до уникального анализа, основанного на водородно-дейтериевом обмене. Тем не менее наиболее надежным представляется метод пептидных карт, обязательный для подтверждения подлинности моноклональных антител для медицинского применения (ОФС.1.7.1.0014.182).
При пептидном картировании исследуемые белки или стандартный и испытуемый образцы специфически расщепляются химически (например, с использованием BrCN) или ферментативно с помощью протеазы, чаще всего трипсина. После этого хроматографически или масс-спектрометрически регистрируются сигналы, соответствующие ожидаемым пептидам [3][4]. Надежность анализа обеспечивается одновременной регистрацией нескольких десятков соответствующих характеристических сигналов. Масс-спектрометрия представляется наиболее предпочтительным методом анализа: высокая точность измерения масс, возможность их точного предварительного расчета обеспечивают внутри- и межлабораторную прецизионность.
При контроле качества лекарственных препаратов белковой природы кроме подтверждения подлинности важными параметрами являются определение общей концентрации белка и его активности. Для определения концентрации белков в растворах наиболее простыми и распространенными методами считаются спектрофотометрические (колориметрические). При этом необходимо, чтобы продукт и субстрат имели различающиеся спектры поглощения или флуоресценции. Если требуется, субстрат модифицируют введением специальных флуоресцирующих или поглощающих групп или используют метки, селективно связывающиеся с субстратом или продуктом. Измерение активности и кинетических констант сводится к определению концентрации: в процессе реакции регистрируется изменение поглощения/флуоресценции субстрата или продукта. Очевидно, что при использовании модифицированных субстратов определяются кинетические параметры реакции с дериватом (производным), а не с нативной молекулой. Такие модификации могут серьезно влиять на значение кинетических параметров. Например, представленные в крупнейшей базе данных ферментов BRENDA3 КМ (константа Михаэлиса) и Ккат (каталитическая константа) трипсина на несколько порядков отличаются для разных субстратов: от 0,15 мкМ до 35,5 мМ [5][6]. Но даже незначительные модификации субстрата, например изомеризация одной аминокислоты, могут в несколько раз изменить скорость реакции [7].
Активность ферментов чаще всего определяют, используя доступные синтетические хромогенные субстраты, по химической природе существенно отличающиеся от природных субстратов. Например, определение активности трипсина согласно ГФ РФ4 сводится к установлению его амидазной активности с субстратом N-α-бензоил-L-аргинин-4-нитроанилидом (BAPNA) [8][9] или, в соответствии с требованиями Фармакопеи США5, эстеразной активности с использованием хромогенного субстрата N-α-бензоил-L-аргинин-этилового эфира (BAEE) [10] (рис. 1). Особенностью спектрофотометрических методов является неселективность, что подразумевает невозможность определения концентрации отдельного компонента сложных смесей, например одного из пептидов, образующихся в результате гидролиза протеазой. Как правило, упомянутые методы требуют использования стандартного образца, или его наличие весьма желательно.
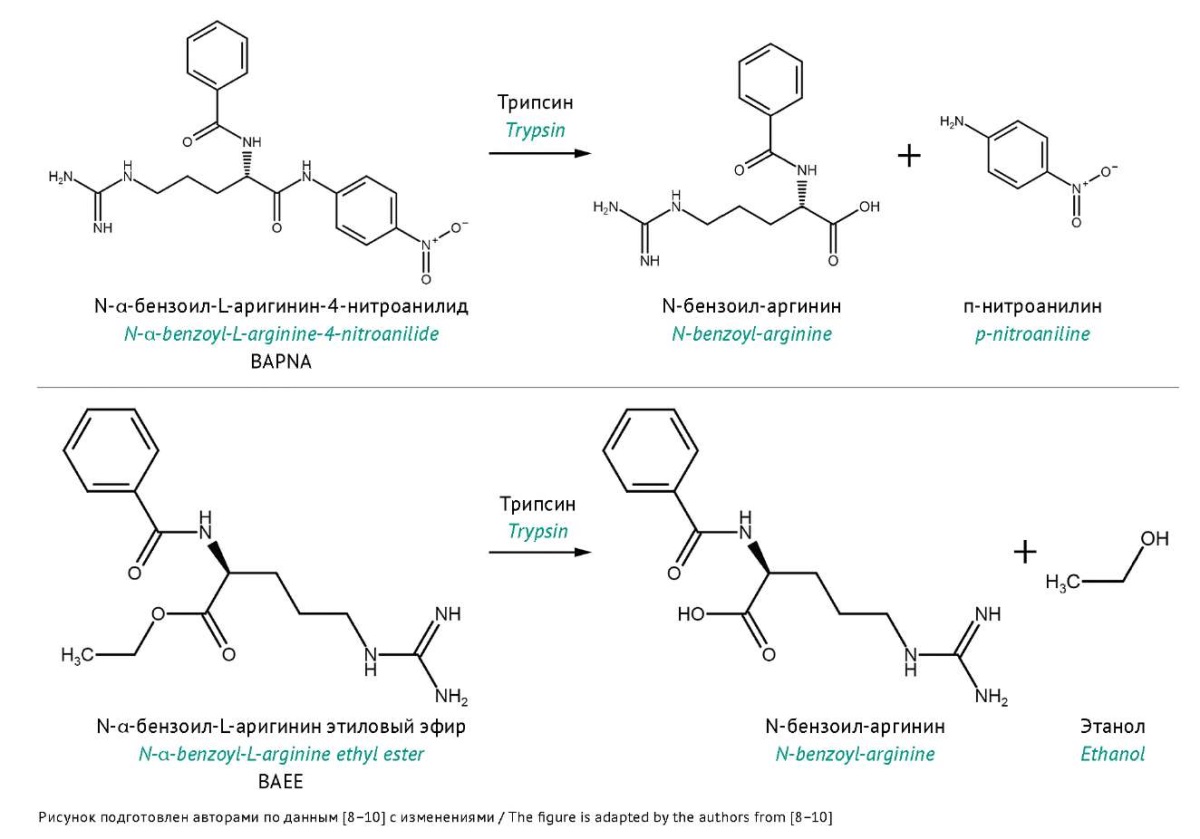
Рис. 1. Субстраты, используемые для определения активности трипсина по продуктам,
детектируемым спектрофотометрически:
BAPNA (с образованием п-нитроанилина) и BAEE (с образованием N-бензоил-аргинина).
Fig. 1. Substrates used in trypsin activity assays,
by products detected by spectrophotometry:
BAPNA (with the formation of p-nitroaniline) and BAEE (with the formation of N-benzoyl-arginine).
Причина недостатков спектрофотометрических методов заключается в том, что при определении концентрации белков и пептидов измеряется концентрация отдельных функциональных групп: заместителей (гетероцикла тирозина и/или триптофана, содержащего ароматическую группу) или аминогрупп, модифицированных радикалами, способными поглощать излучение при определенной длине волны. Суммарно это может приводить к серьезным ошибкам.
Появившиеся в 1980-х гг. биосенсорные методы на основе эффекта поверхностного плазмонного резонанса [11] (surface plasmon resonance, SPR) и электрохимические методы c аффинными модификациями электродов используют для определения кинетических характеристик различных процессов, например констант ассоциации/диссоциации лиганд-рецепторных взаимодействий, кинетики цитохромов Р450 [12] и др. Эти методы предполагают иммобилизацию молекул одного из компонентов реакции на поверхности ячейки SPR или электрода. В результате определяются характеристики гетерофазного процесса. Полученные константы оказываются несопоставимыми с константами реакций в растворе — различными оказываются даже размерности констант (M×см-2 и М×мл-1 для KМ). Ограничением электрохимических методов является необходимость присутствия электроноакцепторной группы в составе иммобилизованной молекулы [13].
Масс-спектрометрические измерения свободны от большинства перечисленных недостатков, но количественные измерения требуют дополнительных манипуляций. При выполнении масс-спектрометрического анализа на некоторые особенности метода следует обратить особое внимание. Прежде всего в масс-спектрометрии измеряется отношение массы к заряду ионов. Это значит, что в масс-спектрах регистрируются сигналы только от ионов. Образование ионов, несущих заряд производных молекул, может происходить по-разному — в результате утраты или присоединения электрона, протона анионного или катионного аддукта, например катиона натрия. Сродство различных соединений к протону или другому агенту, очевидно, не одинаково и может весьма сильно варьироваться. В результате ионизация сложных смесей сопровождается конкуренцией за заряд, а эффективность ионизации компонентов смеси оказывается зависящей от ее состава. Кроме того, в смеси могут оказаться компоненты, гасящие ионизацию, например соли или детергенты.
Таким образом, в смеси, состоящей из нескольких пептидов, интенсивности их сигналов могут меняться от спектра к спектру вне зависимости от их концентрации в образце. Следовательно, исключительно по интенсивностям пиков в масс-спектре без образца сравнения можно только предположительно оценивать концентрации анализируемых веществ.
Это в полной мере относится к так называемым безметочным методам (label-free quantification, LFQ). Например, для оценки относительной концентрации гидролизованных белков используется индекс содержания белка (protein abundance index, PAI) [14], который представляет собой количество идентифицированных пептидов, деленное на количество теоретически наблюдаемых пептидов, или индекс PAI в экспоненциальном представлении — emPAI [15].
В масс-спектрах достаточно высокого разрешения, которые можно получить практически на любом современном масс-спектрометре, органические молекулы, в нашем случае пептиды, представлены несколькими сигналами, называемыми изотопными, которые отличаются для однозарядных ионов на 1 Да. Это следствие существования изотопов — атомов тех же элементов, но отличных по числу нейтронов в ядре. Поэтому в спектре высокого разрешения каждое соединение представлено несколькими сигналами.
Важно, что различающиеся по изотопному составу молекулы химически идентичны, и, независимо от условий регистрации спектров, в том числе состава смеси, соотношение интенсивностей изотопных сигналов не меняется. При этом изотопный состав можно изменить искусственно, заменив часть или все атомы какого-либо элемента на изотопы — ввести изотопную метку. Таким образом получают изотопно-меченый стандарт (ИМС), образец сравнения. В смеси исследуемого образца с природным изотопным распределением и ИМС соотношения изотопных пиков зависят только от того, в каких соотношениях смешаны образец и ИМС. Использование таких изотопных меток (stable isotope labelling, SIL) позволяет с высокой точностью, не хуже спектрофотометрических методов, измерять концентрации по интенсивностям масс-спектрометрических сигналов.
Можно выделить четыре группы методов введения изотопной метки в пептиды и белки:
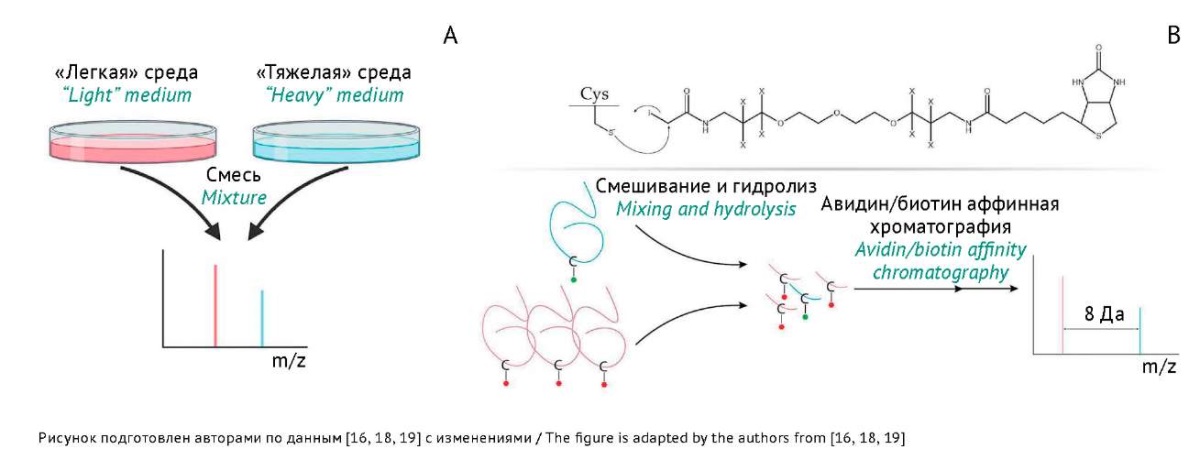
Рис. 2. Методы изотопного мечения для количественного масс-спектрометрического анализа.
A — схема метаболического мечения: синим цветом — клеточная культура на обычной среде («легкая» среда); красным цветом — среда с тяжелыми изотопами («тяжелая» среда). При сравнении двух белков, экстрагированных из клеточных культур, концентрации белков можно определить, сравнивая площади/интенсивности пиков в масс-спектре.
B — тиоловые группы цистеинов модифицируются легкой (X — ¹H) или тяжелой (X — ²H) метками (isotope-coded affinity tag, ICAT). Концентрации белков определяются путем сравнения площади/интенсивности пиков в масс-спектре.
Fig. 2. Isotope-labelling methods for quantitative mass-spectrometric analysis.
A, metabolic labelling scheme: blue, cell growth on a conventional medium (“light” medium); red, cell growth on a medium with heavy isotopes (“heavy” medium). When comparing two proteins extracted from cell cultures, an analyst can determine protein concentrations by comparing peak areas/intensities in the mass spectrum.
B, labelling of cysteine thiol groups with light (X, ¹H) or heavy (X, ²H) isotope-coded affinity tags. Protein concentrations can be determined by comparing peak areas/intensities in the mass spectrum.
Первые три подхода введения изотопной метки довольно дороги и трудозатратны. Использование метода изотопного обмена позволяет получить ИМС за несколько часов и без использования редких реагентов. Изотопный обмен представляет собой спонтанный процесс выравнивания распределения изотопов в различных компонентах системы (например, между растворителем и растворенным соединением). Скорость обмена, очевидно, зависит от того, какие атомы обмениваются, и от положения этих атомов в молекуле. В нашем случае обмен происходит между водой, обогащенной ¹⁸O, и кислородом в пептиде. Наиболее легко обмениваются атомы кислородов карбоксильных групп как C-концевой, так и глутаминовой и аспарагиновой аминокислот. Так как атомы кислорода карбоксильной группы равноправны, то в каждой карбоксильной группе могут обмениваться оба атома кислорода. Изотопный обмен кислорода карбоксильных групп катализируется кислотами (кислотный катализ), а также гидролазами (например, трипсином). При катализе специфичными протеазами обмен преимущественно происходит по сайтам специфического гидролиза. Важно подчеркнуть, что трипсин катализирует обмен кислорода также в уже свободной карбоксильной группе.
Таким образом, ИМС может быть получен в результате гидролиза белка в воде или обменом отдельного пептида в воде H2¹⁸O. А для получения ИМС можно использовать любой уже имеющийся пептид, идентичный измеряемому, например синтетический.
Получение ИМС путем обмена ¹⁶О на ¹⁸O в молекуле пептида не требует дорогостоящих материалов и реактивов или специального синтеза пептидов. Такой ИМС можно использовать для количественного определения пептидов масс-спектрометрически. Определяя концентрацию образовавшегося через некоторое время пептида при разных концентрациях субстрата, можно масс-спектрометрически устанавливать кинетические параметры протеолитических реакций. Предложенный подход не только является альтернативой существующим методам, но и позволяет решить проблему определения активности протеаз, для которых сложно или невозможно получить специальные субстраты.
Цель работы — на примере трипсина и казеина исследовать возможность определения активности фермента методом количественной масс-спектрометрии с меткой ¹⁸O одновременно с подтверждением подлинности.
Реактивы и материалы
В качестве фермента использовали трипсин (Promega, США, кат. № v5111), а в качестве субстрата казеин (Sigma-Aldrich, США, кат. № 9005-46-3). Гидролиз проводили в бикарбонатном буферном растворе pH 7,8 (Sigma-Aldrich, США, кат. № 1066-33-7). Для очистки смеси от казеина и трипсина после гидролиза использовали центрифужные фильтры Microcon 3 кДа (Merсk Millipore, США, кат. № 42404). Обмен атомов кислорода в пептидах проводили в 10% трифторуксусной кислоте, ТФУ (Merсk, США, кат. № 1.08262.0025) в воде H2¹⁸O (НПО «Изотоп», Россия). В качестве матрицы использовали 2,5-дигидроксибензойную кислоту, DHB (Bruker Daltonics, Германия, кат. № 8201346). В качестве подвижных фаз ВЭЖХ применяли ацетонитрил (Merck, кат. № 1.00030.2500) и воду (Merck, кат. № 1.15333.2500).
Оборудование
Разделение пептидов проводили с использованием ВЭЖХ с помощью хроматографа Agilent 1100 (Agilent, США), оснащенного УФ-детектором на диодной матрице и коллектором фракций. Полученные фракции с пептидами высушивали на центрифужном концентраторе Concentrator plus (Eppendorf, Германия).
Масс-спектры получали на времяпролетном масс-спектрометре MALDI-TOF/TOF Bruker Ultraflex II (Bruker Daltonics, Германия), оборудованном Nd:YAG лазером с длиной волны 355 нм, системами задержки экстракции и тандемным селектором LIFT. Спектры регистрировали в положительных ионах в рефлекторном режиме. Каждый спектр получен суммированием серии сигналов, регистрируемых при 1000 импульсах лазера. Масс-спектры фрагментации пептидов регистрировали в тандемном режиме прибора.
Методы
Получение пептидов казеина. Пептиды для введения в них изотопной метки ¹⁸O и дальнейшего использования в качестве ИМС получали гидролизом казеина (15,5 мкМ) трипсином (1,84 мкМ) в 100 мМ аммоний бикарбонатном буферном растворе pH 7,8. Гидролиз проводили в течение 30 мин при 37 °С. Пептидную фракцию полученной смеси отделяли центрифугированием с помощью Microcon 3 кДа и далее разделяли методом обращенно-фазовой ВЭЖХ на колонке Luna® C18(2) (5 мкм, 100 Å, 150×4,6 мм, Phenomenex, США, кат. № 00F-4252-E0). Фаза А — 0,1% ТФУ; фаза В — 0,1% ТФУ, растворенная в ацетонитриле; линейный градиент от 4 до 75% фазы B за 40 мин, поток 1 мл/мин, температура колонки 40 °С. Хроматограммы регистрировали при длинах волн 215 и 280 нм. Собранные на коллекторе фракций пептиды идентифицировали с использованием тандемной масс-спектрометрии MALDI TOF/TOF и высушивали на центрифужном концентраторе до остаточного объема 5 мкл. Концентрации пептидов были рассчитаны по площадям хроматографических пиков; спектрофотометрическая ячейка хроматографа предварительно откалибрована по пептидам, содержащим тирозин и/или триптофан.
Изотопный обмен. В качестве ИМС выбрали триптический пептид казеина TTMPLW c массой 747,3 Да. Для введения изотопной метки высушенные пептиды растворяли в 10% ТФУ в H2¹⁸O и инкубировали 2 ч при 60 °С.
Гидролиз белка для определения кинетики трипсина. В качестве субстрата был использован раствор казеина с концентрациями 7,75, 3,87 и 3,1 мкМ. Смешивали 16 мкл раствора казеина и 12 мкл раствора трипсина (концентрация 0,02 мг/мл) в аммоний-бикарбонатном буферном растворе pH 7,8. Смесь инкубировали в течение 15 мин.
Масс-спектрометрия MALDI. В качестве матрицы использовали 10 мкг/мл DHB, растворенной в смеси 50% ацетонитрила с 0,4% ТФУ. 0,5 мкл образца смешивали с 0,5 мкл раствора матрицы непосредственно на масс-спектрометрической мишени и высушивали на воздухе.
Количественный анализ MALDI. Для количественного масс-спектрометрического анализа от 3 до 10 мкл растворов пептидов или реакционных смесей гидролиза казеина трипсином смешивали с пептидами с внедренной меткой ¹⁸O в соотношениях 1:2, 1:1, 2:1 и перемешивали. Сразу после этого к полученным смесям добавляли раствор матрицы и быстро наносили на масс-спектрометрическую мишень и высушивали на воздухе.
В качестве матрицы использовали DHB. Для каждой пробы было получено не менее трех спектров.
Идентификация казеина и трипсина
Смесь продуктов гидролиза казеина трипсином содержала фрагменты, соответствующие четырем цепям этого белка. Перекрытие аминокислотной последовательности α-S1 казеина составило 48%, α-S2 казеина — 38%, β-казеина — 21% и каппа-казеина (κ-казеина) — 28%. Пептиды автолиза трипсина перекрывали 38% его последовательности. Не все пептиды находятся в диапазоне, регистрируемом масс-спектрометрически. Если необходимо полное покрытие, используют несколько протеаз [21]. Однако такого процента покрытия может быть достаточно для подтверждения подлинности одновременно белка и фермента (особенно если комбинировать такой метод с методом тандемной масс-спектрометрии), а также подтверждения специфичности фермента.
Получение изотопно-меченого стандарта и определение его концентрации
Количественный масс-спектрометрический анализ с использованием изотопных меток позволяет определить отношение концентраций изучаемого образца к концентрации ИМС. То есть для определения концентрации изучаемого пептида необходимо и достаточно знать концентрацию ИМС. Метод определения концентрации ИМС не имеет значения, как и происхождение пептида. Важным является его химическая идентичность измеряемому образцу. Пептид может быть синтезирован химически или получен путем гидролиза субстрата и последующего выделения его продуктов на ВЭЖХ. В настоящей работе использовали второй подход, так как по мнению авторов он является более простым и удобным.
Концентрацию ИМС определяли, смешивая его с таким же природным пептидом без изотопной метки (эталоном) с известной концентрацией. Концентрацию эталона определяли по площади хроматографического пика. Зависимость концентрации от поглощения при 280 нм получена с использованием модельных триптофан-содержащих пептидов.
Концентрацию небольших пептидов, содержащих одну или несколько ароматических аминокислот, можно с высокой точностью вычислить по поглощению при длине волны 280 нм. Коэффициенты экстинкции тирозина и триптофана известны [22], при этом поглощение пептидных связей и радикалов других аминокислот при этой длине волны пренебрежимо мало. Это позволяет вычислять концентрации по площадям хроматографических пиков. Концентрация эталонного пептида без изотопной метки составила 26 мкМ (±2мкМ).
Следует отметить, что если задачи эксперимента позволяют выбрать пептид для измерений, то желательно избежать возможного наложения спектра с другими пептидами близкой массы. Это связано с тем, что при кислотном катализе обмениваться будут атомы кислорода всех карбоксильных групп, а значит, спектр пептида будет расширяться в сторону бóльших масс.
В данной работе был выбран пептид TTMPLW (рис. 3), не содержащий аминокислот, в радикалах которых имеются карбоксильные группы, то есть аспарагиновой и глутаминовой кислот.
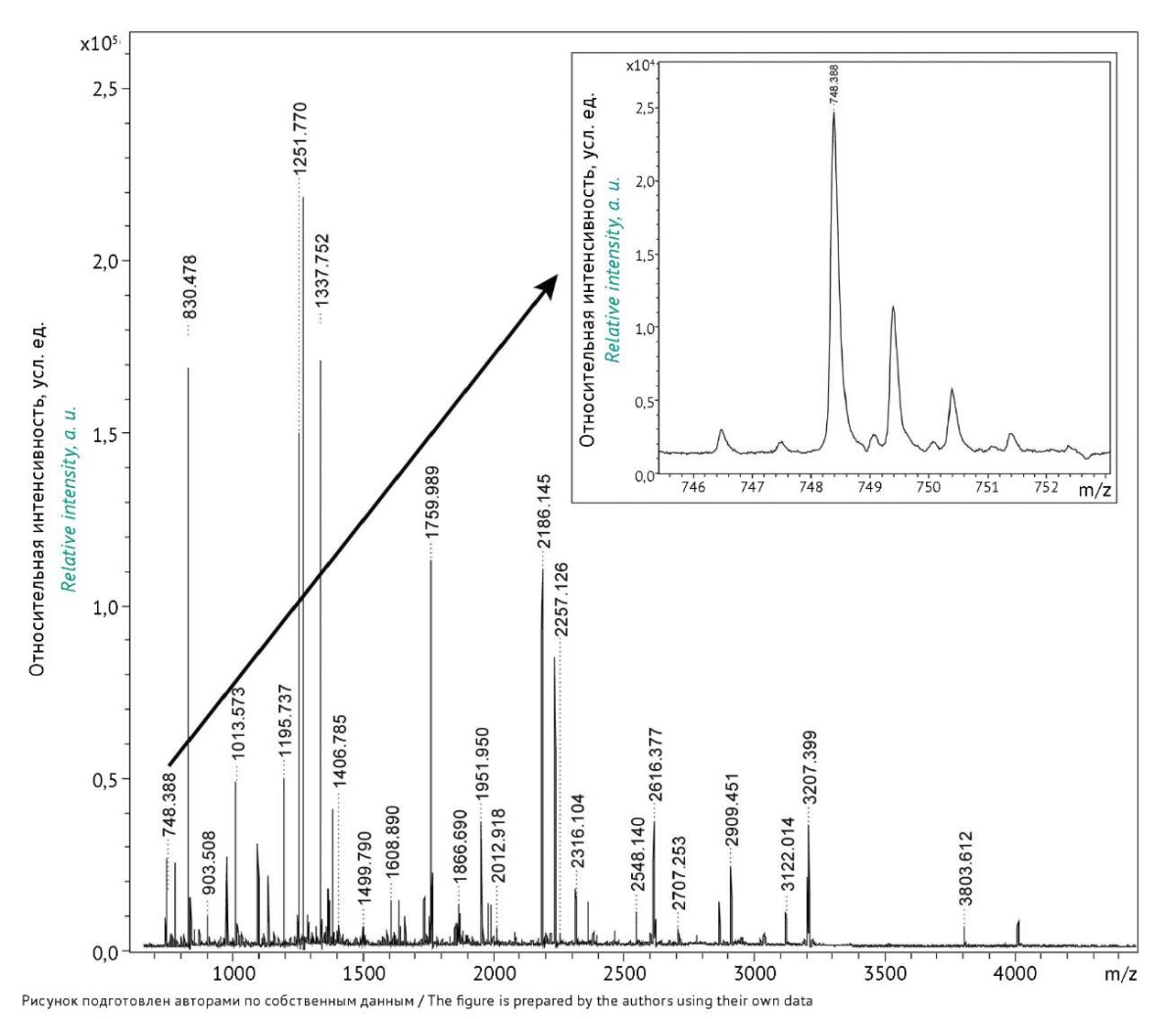
Рис. 3. Масс-спектр смеси продуктов трипсинолиза казеина.
С правой стороны представлен масс-спектр пептида TTMPLW
(молекулярная масса 747,3 Да) в увеличенном масштабе.
m/z — отношение массы заряженных частиц (ионов) к их заряду.
Fig. 3. Mass spectrum of tryptic casein hydrolysates.
The insert in the right upper corner shows the mass spectrum
of the TTMPLW peptide (molecular weight: 747.3 Da) in a larger scale.
m/z is the mass-to-charge ratio of charged particles (ions).
Для контроля обмена атомов ¹⁶О на изотопы ¹⁸O в пептиде в течение трех часов были получены спектры (рис. 4). После одного часа инкубации замена атомов ¹⁶О на их изотопы ¹⁸O прошла не полностью, но через два часа площадь первого пика составила менее 1% площади пятого пика. Сравнивая площади пиков изотопных сигналов, соответствующих пептидам с одним или двумя включенными (обменянными) кислородами, можно определить, что в 18% молекул обменялся один атом кислорода, в 81% — два атома и в ~1% молекул не обменялось ни одного атома кислорода. Важно, что для количественной оценки пептида не обязательно использовать ИМС, в котором замена ¹⁶О на ¹⁸O произошла во всех молекулах пептида или полнота обмена была близкой к таковой. Полученные результаты позволили далее вычислять концентрацию исследуемого пептида, зная только концентрации ИМС и соотношения площадей изотопных пиков в масс-спектрах образцов природного пептида.
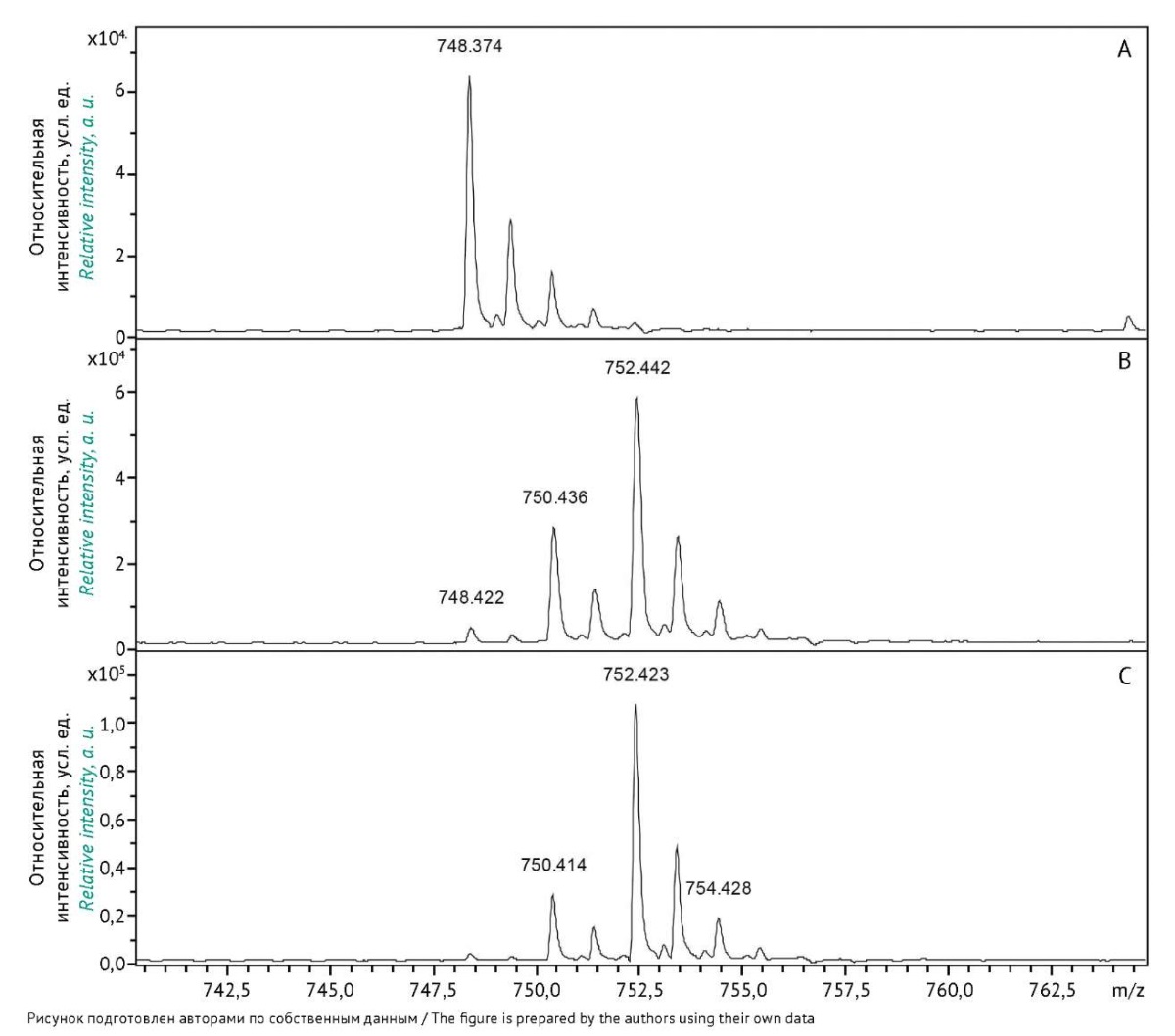
Рис. 4. Масс-спектры пептида TTMPLW после его перерастворения
в 10% растворе трифторуксусной кислоты в H2¹⁸O
через 5 мин (A), 60 мин (B) и 120 мин (C) инкубации.
Fig. 4. Mass spectra of the TTMPLW peptide redissolved
in 10% trifluoroacetic acid in H2¹⁸O and incubated
for 5 minutes (A), 60 minutes (B), and 120 minutes (C).
Методом тандемной масс-спектрометрии подтверждено, что в карбоксильной группе С-концевого триптофана в пептиде TTMPLW два атома ¹⁶О заместились на два атома ¹⁸O (рис. 5). Наблюдаемые N-концевые фрагменты исходного пептида и пептида после обмена идентичны, в то время как массы С-концевых фрагментов отличаются на 4 Да, что соответствует обмену двух атомов кислорода карбоксильной группы на ¹⁸O. Несмотря на то что в пептиде имеется остаток треонина, который содержит атом кислорода в гидроксильной группе радикала, потенциально способный к обмену, этого не наблюдается, так как скорости обмена атомов разных функциональных групп различаются на несколько порядков.
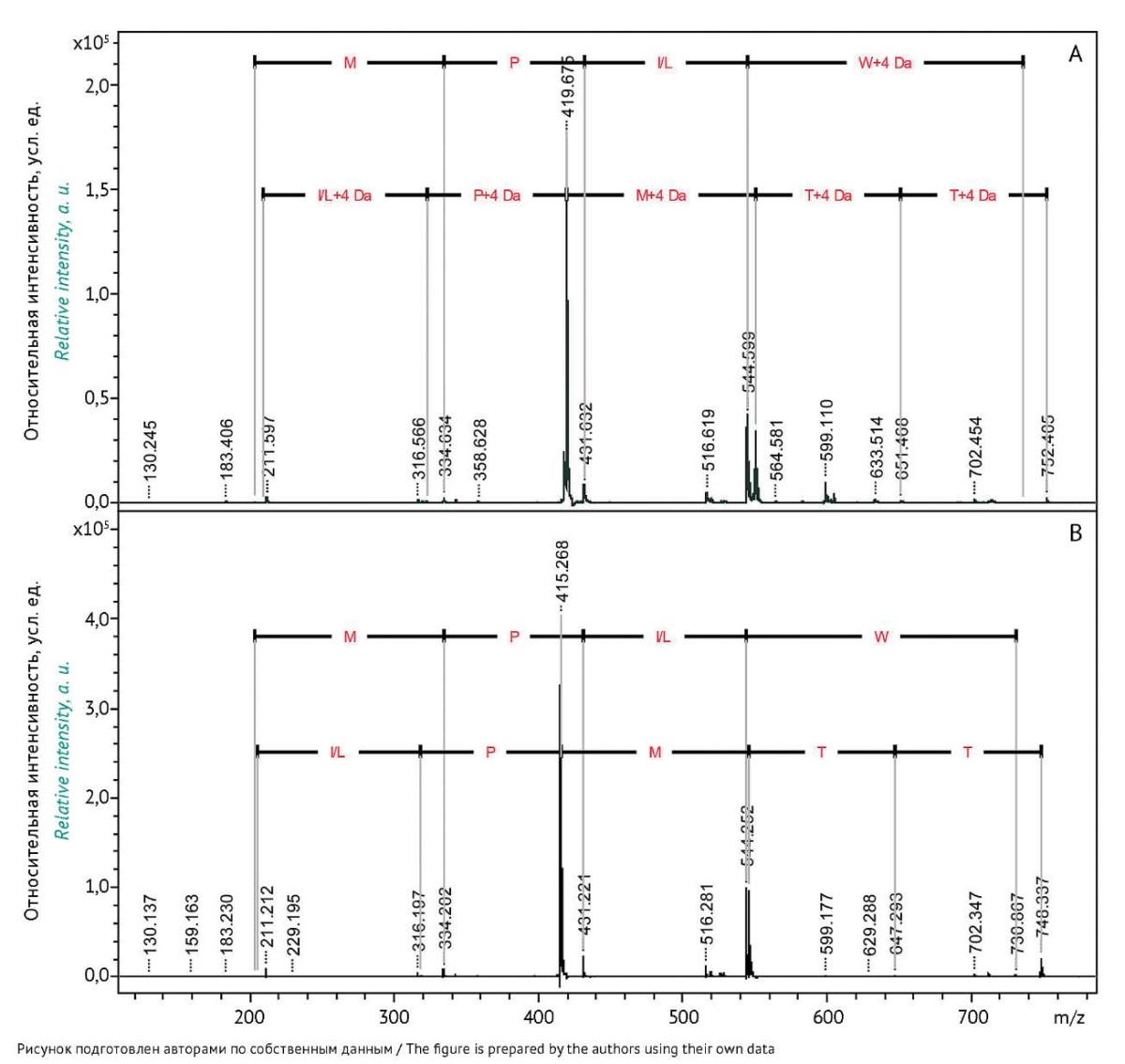
Рис. 5. Масс-спектры фрагментов пептида TTMPLW
после инкубации в воде H2¹⁸O (А) и исходного пептида (В).
Fig. 5. Mass spectra of TTMPLW peptide fragments
incubated in H2¹⁸O (А) and the intact peptide (В).
Расчет концентрации пептида по изотопно-меченому стандарту
Для определения концентрации полученного ИМС его смешивали с эталонным пептидом известной концентрации, вычисленной по площади хроматографического пика. ИМС и пептид TTMPLW (продукт или эталон) желательно смешивать в таком соотношении, чтобы интенсивности моноизотопного пика продукта и наиболее интенсивного пика ИМС с включенными ¹⁸O были примерно одинаковыми. В нашем случае оптимальное соотношение продукта к ИМС составило 2:1.
Следует отметить, что при изотопном обмене сложно добиться замены только одного или только двух атомов кислорода. В результате получившийся ИМС содержит смесь всех трех изотопных форм пептида, которые накладываются в спектре: пептиды без ¹⁸O, с одним изотопом ¹⁸O и пептиды с двумя ¹⁸O (рис. 4B и 6В). В части молекул произошла замена только одного атома кислорода карбоксильной группы на ¹⁸O и привела к тому, что изотопные пики этого пептида сдвинулись на 2 атомные единицы массы (а.е.м.) вправо (рис. 6B, выделено желтым цветом). При этом соотношение интенсивностей изотопных пиков этого пептида сохраняется. В другой части молекул произошла замена обоих атомов кислорода карбоксильной группы, которая привела к тому, что изотопные пики этого пептида сдвинулись на 4 а.е.м. (рис. 6B, выделено зеленым цветом).

Рис. 6. Схематическое изображение масс-спектров.
A — пептид TTMPLW (продукт);
B — изотопно-меченый стандарт (ИМС) с заменой одного или двух атомов ¹⁸O;
C — смеси ИМС и продукта, где наглядно показано
наложение относительных интенсивностей пиков ИМС и продукта.
Химическими формулами изображены атомы кислорода,
способные обмениваться в исследуемом пептиде.
Fig. 6. Schematic mass spectra:
A, TTMPLW peptide (product);
B, isotope-labelled standard peptide with one and two ¹⁸O exchanges;
C, mixtures of the standard peptide and the product,
clearly showing the superposition of the relative intensities of the respective peaks.
The chemical formulas depict the oxygen atoms in the peptide that are capable of exchange.
Обычно удается получить ИМС, в котором практически отсутствуют пики пептида без включения ¹⁸O. В этом случае отношение концентраций пептида-продукта и ИМС будет равно отношению интенсивности первого пика к сумме третьего и пятого пиков (рис. 6B). Это пики ИМС, но на них накладываются третий и пятый пик продукта, а на пятый пик (пептиды с двумя включенными изотопами ¹⁸O) — еще и третий пик пептидов (пептиды с одним включенным изотопом ¹⁸O). Вклады этих пептидов нужно вычесть [23] согласно формуле (1):
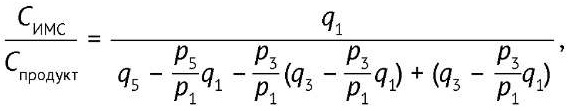 (1)
(1)
где q1, q3 и q5 — интенсивности первого, третьего и пятого сигналов в спектре смеси продукта реакции и ИМС, p1, p3 и p5 — соответствующие интенсивности сигналов в спектре исходного пептида с неизмененным изотопным составом. Для проведения расчета можно не регистрировать спектр природного пептида, а взять его из другого эксперимента или рассчитать p1, p3 и p5 теоретически.
Следует отметить, что нет необходимости знать соотношение пиков в спектре ИМС, достаточно только знать, что замена атомов ¹⁶О на ¹⁸O произошла в большей части пептидов, а число атомов в молекуле пептида, которые могут подвергаться замене, невелико. Однако распределение в экспериментально полученных пептидах часто оказывается отличным от расчетного, что приводит к неправильному расчету p1, p3, p5 и увеличению ошибок в значении концентрации.
Последние ограничения не исключают возможности точного расчета концентраций: даже если ИМС будет не идеальным, в положении первого пика окажется достаточно сильный сигнал, обмен пройдет не полностью, а подвергаться замене будет большое число атомов, точный расчет концентраций все равно остается возможным — в этом случае нужно будет использовать три спектра:
Интенсивность каждого сигнала в спектре смеси окажется суммой соответствующих сигналов эталонного пептида и ИМС с коэффициентами t и k (2):
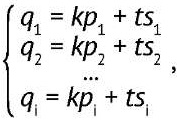 (2)
(2)
где qi — интенсивность сигнала пика i в спектре смеси продукта реакции и ИМС; pi — интенсивность сигнала пика i в спектре эталонного пептида (с неизмененным изотопным составом); si — интенсивность сигнала пика i в спектре ИМС.
В силу того, что в различных опытах (даже повторах) величины qi, pi и si могут сильно меняться, рассчитанные значения коэффициентов t и k также будут изменяться. Однако, как было сказано ранее, если компоненты смеси химически идентичны, то соотношение пиков таких компонентов не меняется. Для определения концентрации пептида продукта необходимо найти отношение коэффициентов t к k, которое сохраняется для разных спектров. Чтобы найти t и k, достаточно двух уравнений; желательно выбрать два наиболее интенсивных сигнала. В нашем случае ими оказались q1 и q5 (3):
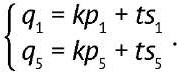 (3)
(3)
Отношение k к t равняется отношению концентрации продукта к концентрации ИМС и находится путем решения системы линейных алгебраических уравнений согласно (4):
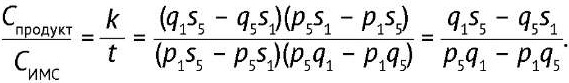 (4)
(4)
Поскольку пятого пика в спектре эталона мы не наблюдали (p5=0), а первый пик эталона и пятый пик ИМС нормируются на 1 (p1=1, s5=1), то уравнение принимает вид (5):
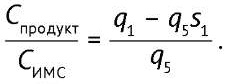 (5)
(5)
Определение концентрации продукта проводили при использовании этого подхода дважды — устанавливая концентрацию ИМС по спектру его смеси с эталоном и затем определяя концентрацию продукта реакции по спектру его смеси с ИМС.
Определение константы Михаэлиса трипсина
В ходе работы проводилось определение количества пептида-продукта, образующегося при гидролизе трех различных концентраций субстрата (казеина) трипсином за 15 мин. Эти данные далее использовались для измерения активности фермента.
Для более точного определения концентрации масс-спектрометрическим методом необходимо смешивать ИМС и продукт в таком соотношении, чтобы относительные интенсивности сигналов этих пептидов на масс-спектре были примерно одинаковыми. Поскольку с уменьшением количества субстрата уменьшается и количество продукта, то к образцам добавлялся ИМС с различным соотношением: для концентрации казеина 7,75 мкМ соотношение субстрата к ИМС было 1:1, а для концентраций казеина 3,87 и 3,1 мкМ — 1:2,5. Несмотря на то, что концентрацию ИМС можно посчитать из одного спектра — смеси ИМС с эталоном — более точным будет измерение концентрации ИМС масс-спектрометрически для каждого разведения (соотношения субстрат-ИМС). Поэтому концентрация ИМС, рассчитанная по спектру смеси ИМС и эталона в соотношении 1:1, составила 11,0±0,2 мкМ, а в случае соотношения 1:2,5 — 3,8 мкМ.
Затем по известной концентрации ИМС была рассчитана концентрация пептида-продукта. Для концентрации казеина 7,75 мкМ она составила 5,00±0,04 мкМ (или 0,3 мкМ/мин), для концентрации казеина 3,87 мкМ — 3,00±0,29 мкМ (или 0,2 мкМ/мин), а для концентрации 3,1 мкМ — 2,55±0,25 мкМ (или 0,17 мкМ/мин). Стандартное отклонение значений повторных измерений показало, что ошибка измерений оказывается меньше по сравнению со спектрофотометрическим методом, а минимальная установленная чувствительность предложенного метода составила 0,50±0,08 мкМ. При этом чувствительность МАЛДИ масс-спектрометрии существенно выше этих значений, что указывает на возможное достижение еще более высокой чувствительности.
По полученным данным строили график зависимости скорости образования пептида-продукта от концентрации субстрата (казеин) в обратных координатах (рис. 7). Как видно из графика, наблюдается зависимость увеличения количества продукта от увеличения концентрации субстрата в обратных координатах и, следовательно, уменьшение концентрации субстрата при уменьшении концентрации фермента в прямых.
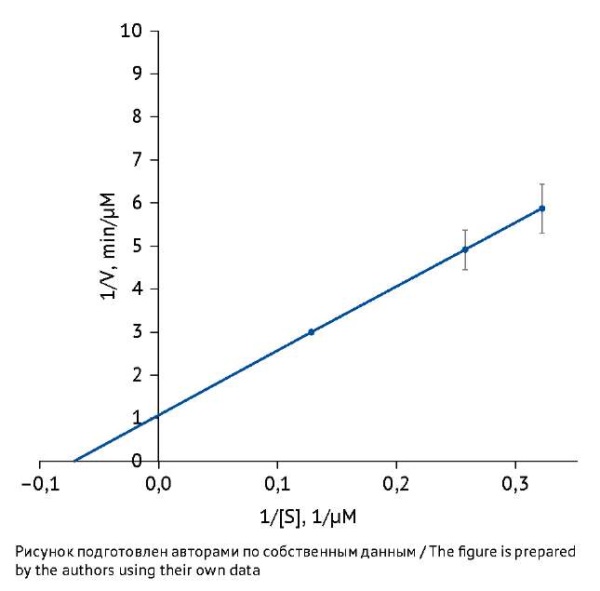
Рис. 7. Зависимость скорости образования пептида-продукта
от концентрации субстрата (казеин) в двойных обратных координатах.
По оси Y — 1/скорость образования продукта (мин/мкМ),
по оси X — 1/концентрация субстрата (1/мкМ).
Fig. 7. Double reciprocal plot of the product formation rate plotted
against the substrate (casein) concentration.
Y-axis, 1/product formation rate (min/MM);
X-axis, 1/substrate concentration (1/MM).
Расчет КМ при использовании описанной методики определения активности фермента показал, что КМ для трипсина составила 13,65±0,60 мкМ. Сравнение полученного значения константы с КМ, доступными в базе данных BRENDA для казеина при сходных температурных условиях и pH, показало, что КМ=3,33 мкМ [24] отличается от полученных нами данных практически в 4 раза. Возможно, это объясняется тем, что в указанной работе казеин был модифицирован меткой FITC, которая является радикалом, реагирующим с первичными аминами белков по остаткам лизина и на аминогруппе белка. Как следствие данной модификации, такой субстрат отличается от использованного нами, и, следовательно, скорость гидролиза трипсина по такому субстрату должна отличаться. Следует добавить, что активность самого фермента может отличаться для разных партий фермента даже одного производителя6, что также влияет на кинетические параметры.
Представленный метод введения изотопной метки является универсальным и может быть использован для любой протеазы, поскольку образующиеся в результате гидролиза пептиды имеют как минимум два атома кислорода на С-концевой карбоксильной группе, способные к обмену на изотоп ¹⁸O. Также для каждой протеазы можно подобрать пептид, по которому можно проводить расчет, однако такой пептид должен отвечать ряду требований. Поскольку сигналы ИМС в спектре находятся справа (соответствуют большим значениям m/z) от сигналов измеряемого пептида, желательно, чтобы в этом участке спектра отсутствовали сигналы других ионов, которые могут накладываться на сигналы ИМС. Кроме того, предложенный метод не требует специальных реактивов (например, BAEE и BAPNA) и растворителей, а в качестве субстрата можно использовать любой белок. Вода, обогащенная ¹⁸O, применяется в медицинской диагностике, производится в достаточно больших объемах и не требует больших затрат.
Следует отметить, что предложенный авторами в данной работе метод является ресурсозатратным, и для него необходимо наличие масс-спектрометра и хроматографа. К недостаткам также можно отнести этап подбора перед анализом пептида, по которому будут измеряться кинетические параметры.
Вклад авторов. Все авторы подтверждают соответствие своего авторства критериям ICMJE. Наибольший вклад распределен следующим образом: М.А. Константинов и И.Ю. Торопыгин — формирование концепции и дизайна работы, проведение масс-спектрометрического анализа, разделение пептидов с использованием ВЭЖХ, обработка данных, написание и редактирование текста рукописи; Д.Д. Жданов — формирование концепции, написание текста рукописи.
Благодарности. Выражаем благодарность Е.В. Ивановой за помощь в подготовке рукописи.
Authors’ contributions. All the authors confirm that they meet the ICMJE criteria for authorship. The most significant contributions were as follows. M.A. Konstantinov and I.Yu. Toropygin conceptualised and designed the study, carried out mass-spectrometric analysis and HPLC peptide separation, processed data, drafted and edited the manuscript. D.D. Zhdanov conceptualised the study and drafted the manuscript.
Acknowledgements. The authors thank E.V. Ivanova for her assistance in preparing the manuscript.
1. ОФС.1.7.2.0035.18 Пептидное картирование. Государственная фармакопея Российской Федерации. XIV изд. Т. 2; 2018.
ОФС.1.7.1.0007.15 Лекарственные средства, получаемые методами рекомбинантных ДНК. Государственная фармакопея Российской Федерации. XIV изд. Т. 2; 2018.
ОФС.1.7.1.0014.18 Моноклональные антитела для медицинского применения. Государственная фармакопея Российской Федерации. XIV изд. Т. 2; 2018.
2. Там же.
3. https://www.brenda-enzymes.org/
4. ФС.2.1.0195.18 Трипсин. Государственная фармакопея Российской Федерации. XIV изд. Т. 3; 2018.
5. USP 43–NF 38 <89> Enzymes used as ancillary materials in pharmaceutical manufacturing.
6. https://www.promega.com/-/media/files/resources/protocols/product-information-sheets/n/sequencing-grade-modified-trypsin-protocol.pdf?la=en
1. Lukasheva EV, Babayeva G, Karshieva SS, Zhdanov DD, Pokrovsky VS. L-lysine α-oxidase: enzyme with anticancer properties. Pharmaceuticals (Basel). 2021;14(11):1070. https://doi.org/10.3390/ph14111070
2. Pokrovskaya MV, Pokrovsky VS, Aleksandrova SS, Sokolov NN, Zhdanov DD. Molecular analysis of L-asparaginases for clarification of the mechanism of action and optimization of pharmacological functions. Pharmaceutics. 2022;14(3):599. https://doi.org/10.3390/pharmaceutics14030599
3. Liska AJ, Shevchenko A. Combining mass spectrometry with database interrogation strategies in proteomics. TrAC Trends Anal Chem. 2003;22(5):291–8. https://doi.org/10.1016/S0165-9936(03)00507-7
4. Голощапова ЕО, Рунова ОБ, Минеро АС, Фадейкина ОВ, Волкова РА, Дегтерев МБ и др. Разработка и аттестация фармакопейного стандартного образца для подтверждения подлинности первичной структуры очищенного рекомбинантного интерферона бета-1b методом пептидного картирования. БИОпрепараты Профилактика, диагностика, лечение. 2022;22(1):23–37. https://doi.org/10.30895/2221-996X-2022-22-1-23-37
5. Zhang Y, Ling Z, Du G, Chen J, Kang Z. Improved production of active Streptomyces griseus trypsin with a novel auto-catalyzed strategy. Sci Rep. 2016;6(1):23158. https://doi.org/10.1038/srep23158
6. Huang CT. Vertebrate serum inhibitors of Aedes aegypti trypsin. Insect Biochem. 1971;1(1):27–38. https://doi.org/10.1016/0020-1790(71)90019-9
7. Toropygin IYu, Kugaevskaya EV, Mirgorodskaya OA, Elisseeva YuE, Kozmin YuP, Popov IA, et al. The N-domain of angiotensin-converting enzyme specifically hydrolyzes the Arg-5-His-6 bond of Alzheimer’s Aβ-(1-16) peptide and its isoAsp-7 analogue with different efficiency as evidenced by quantitative matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2008;22(2):231–9. https://doi.org/10.1002/rcm.3357
8. Li M, Rauf A, Guo Y, Kang X. Real-time label-free kinetics monitoring of trypsin-catalyzed ester hydrolysis by a nanopore sensor. ACS Sens. 2019;4(11):2854–7. https://doi.org/10.1021/acssensors.9b01783
9. Treetharnmathurot B, Ovartlarnporn C, Wungsintaweekul J, Duncan R, Wiwattanapatapee R. Effect of PEG molecular weight and linking chemistry on the biological activity and thermal stability of PEGylated trypsin. Int J Pharm. 2008;357(1–2):252–9. https://doi.org/10.1016/j.ijpharm.2008.01.016
10. Senphan T, Benjakul S, Kishimura H. Purification and characterization of trypsin from hepatopancreas of Pacific white shrimp. J Food Biochem. 2015;39(4):388–97. https://doi.org/10.1111/jfbc.12147
11. Homola J, Yee S, Gauglitz G. Surface plasmon resonance sensors: review. Sensors Actuators B Chem. 1999;54(1):3-15. https://doi.org/10.1016/S0925-4005(98)00321-9
12. Shumyantseva VV, Kuzikov AV, Masamrekh RA, Bulko TV, Archakov AI. From electrochemistry to enzyme kinetics of cytochrome P450. Biosens Bioelectron. 2018;121:192–204. https://doi.org/10.1016/j.bios.2018.08.040
13. Filippova TA, Masamrekh RA, Shumyantseva VV, Latsis IA, Farafonova TE, Ilina IY, et al. Electrochemical biosensor for trypsin activity assay based on cleavage of immobilized tyrosine-containing peptide. Talanta. 2023;257:124341. https://doi.org/10.1016/j.talanta.2023.124341
14. Rappsilber J, Ryder U, Lamond AI, Mann M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002;12(8):1231–45. https://doi.org/10.1101/gr.473902
15. Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4(9):1265–72. https://doi.org/10.1074/mcp.M500061-MCP200
16. Chen X, Wei S, Ji Y, Guo X, Yang F. Quantitative proteomics using SILAC: principles, applications, and developments. Proteomics. 2015;15(18):3175–92. https://doi.org/10.1002/pmic.201500108
17. Vogl DP, Conibear AC, Becker CFW. Segmental and site-specific isotope labelling strategies for structural analysis of posttranslationally modified proteins. RSC Chem Biol. 2021;2(5):1441–61. https://doi.org/10.1039/D1CB00045D
18. Chahrour O, Cobice D, Malone J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J Pharm Biomed Anal. 2015;113:2–20. https://doi.org/10.1016/j.jpba.2015.04.013
19. Petriz BA, Franco OL. Application of cutting-edge proteomics technologies for elucidating host-bacteria interactions. Adv Protein Chem Struct Biol. 2014;95:1–24. https://doi.org/10.1016/B978-0-12-800453-1.00001-4
20. Kozmin YP, Manoilov AV, Serebryakova MV, Mirgorodskaya OA. A direct introduction of ¹⁸О isotopes into peptides and proteins for quantitative mass spectroscopy analysis. Russ J Bioorganic Chem. 2011;37(6):719–31. https://doi.org/10.1134/S1068162011060094
21. Swaney DL, Wenger CD, Coon JJ. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J Proteome Res. 2010;9(3):1323–9. https://doi.org/10.1021/pr900863u
22. Mach H, Middaugh CR, Lewis RV. Statistical determination of the average values of the extinction coefficients of tryptophan and tyrosine in native proteins. Anal Biochem. 1992;200(1):74–80. https://doi.org/10.1016/0003-2697(92)90279-G
23. Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic ¹⁸О labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2004;76(9):2675. https://doi.org/10.1021/ac049600x
24. Finehout EJ, Cantor JR, Lee KH. Kinetic characterization of sequencing grade modified trypsin. Proteomics. 2005;5(9):2319–21. https://doi.org/10.1002/pmic.200401268
Константинов Михаил Александрович
ул. Погодинская, д. 10, стр.8, Москва, 119121
Жданов Дмитрий Дмитриевич, д-р биол. наук
ул. Погодинская, д. 10, стр.8, Москва, 119121
Торопыгин Илья Юрьевич, канд. биол. наук
ул. Погодинская, д. 10, стр.8, Москва, 119121
пос. Борок, д. 109, Некоузский район, Ярославская область, 152742
Константинов М.А., Жданов Д.Д., Торопыгин И.Ю. Количественная масс-спектрометрия с меткой ¹⁸О как альтернативный подход к определению активности протеаз на примере трипсина. БИОпрепараты. Профилактика, диагностика, лечение. 2024;24(1):46-60. https://doi.org/10.30895/2221-996X-2024-24-1-46-60
Konstantinov M.A., Zhdanov D.D., Toropygin I.Yu. Quantitative mass spectrometry with ¹⁸O labelling as an alternative approach for determining protease activity: an example of trypsin. Biological Products. Prevention, Diagnosis, Treatment. 2024;24(1):46-60. (In Russ.) https://doi.org/10.30895/2221-996X-2024-24-1-46-60

Издатель: ФГБУ «НЦЭСМП» Минздрава России































