




Содержание
Перейти к:
https://doi.org/10.30895/2221-996X-2025-25-1-71-82
Перейти к:
ВВЕДЕНИЕ. Совершенствование технологий производства, расширение спектра биологических лекарственных препаратов (БЛП) и показаний к их применению обусловливают необходимость формирования единого подхода к их стандартизации в рамках как Государственной фармакопеи Российской Федерации (ГФ РФ), так и региональной фармакопеи — Фармакопеи Евразийского экономического союза (Фармакопея Союза). В условиях единого фармацевтического рынка стран Евразийского экономического союза (ЕАЭС) требования к лекарственным препаратам должны учитывать национальные фармакопейные стандарты качества, которые, в свою очередь, подвергаются пересмотру с целью гармонизации с Фармакопеей Союза.
ЦЕЛЬ. Систематизация требований к биологическим лекарственным препаратам в рамках Фармакопеи Союза и Государственной фармакопеи Российской Федерации для гармонизации региональных и национальных стандартов.
ОБСУЖДЕНИЕ. Проведен анализ особенностей стандартизации БЛП в фармакопеях государств — членов ЕАЭС, а также требований ведущих мировых фармакопей и международных нормативных документов, в том числе Международного совета по гармонизации технических требований к лекарственным средствам для медицинского применения (ICH). Установлены основные принципы формирования требований к БЛП в рамках Фармакопеи Союза и подготовлен проект фармакопейной статьи (ОФС) «Биологические лекарственные препараты». В данной ОФС унифицировано определение понятия «биологические лекарственные препараты» с учетом региональных и российских законодательных актов; проведена фармакопейная стандартизации требований к различным группам БЛП; выделены основные аспекты, требующие рассмотрения в рамках производства (технологии), контроля качества промежуточных продуктов и лекарственного препарата. В процессе гармонизации ГФ РФ с требованиями Фармакопеи Союза представлены основные подходы к стандартизации БЛП в ГФ РФ и внесены изменения в ОФС.1.7.1.0010.18 «Биологические лекарственные препараты», в том числе в части исключения требований к производству, которые не являются общими для всех БЛП, а также исключения перечня испытаний фармацевтических субстанций и лекарственных препаратов.
ЗАКЛЮЧЕНИЕ. Систематизированы требования к БЛП с учетом мировых и региональных нормативных документов. Подготовлен проект ОФС «Биологические лекарственные препараты» для Фармакопеи Союза и на его основе актуализирована ОФС.1.7.1.0010.18 «Биологические лекарственные препараты». Обоснованы подходы к формированию фармакопейных статей и общих фармакопейных статей, касающихся требований к различным группам БЛП.
Корнилова О.Г., Багирова В.Л. Фармакопейная стандартизация биологических лекарственных препаратов: основные принципы в условиях единого фармацевтического рынка стран Евразийского экономического союза. БИОпрепараты. Профилактика, диагностика, лечение. 2025;25(1):71-82. https://doi.org/10.30895/2221-996X-2025-25-1-71-82
Kornilova O.G., Bagirova V.L. Pharmacopoeial standardisation of biological medicinal products: Basic principles for the common pharmaceutical market of the Eurasian Economic Union. Biological Products. Prevention, Diagnosis, Treatment. 2025;25(1):71-82. (In Russ.) https://doi.org/10.30895/2221-996X-2025-25-1-71-82
Биологические лекарственные препараты (БЛП) представляют собой разнообразную группу лекарственных препаратов, получаемых из биологических источников. Современные методы лечения, такие как иммунотерапия с использованием моноклональных антител и клеточная терапия, способствовали росту спроса на биотехнологические и высокотехнологичные лекарственные препараты [1][2]. Применение биотехнологических подходов позволяет разрабатывать персонализированные стратегии лечения, основанные на уникальных генетических профилях конкретных пациентов или подтипах заболеваний. Несмотря на высокие производственные затраты и строгие регуляторные требования, мировой рынок БЛП в 2023 г. достиг уровня в 580 млрд долларов США1. Прогнозируемый среднегодовой рост рынка в последующие восемь лет составит 6%2.
Постоянное совершенствование технологий производства, расширение спектра БЛП и показаний к их применению обусловливают необходимость формирования единого подхода к их стандартизации в рамках как национальной фармакопеи — Государственной фармакопеи Российской Федерации (ГФ РФ), так и региональной фармакопеи — Фармакопеи Евразийского экономического союза (Фармакопея Союза) [1][2]. Соглашение о единых принципах и правилах обращения лекарственных средств (ЛС) в рамках Евразийского экономического союза (ЕАЭС), ратифицированное Российской Федерацией в 2016 г.3, предусматривает проведение скоординированной политики в сфере обращения ЛС в первую очередь в целях обеспечения их качества, гарантирующего эффективность и безопасность при применении. Единство требований к качеству ЛС на территории государств — членов ЕАЭС возможно только при наличии единого подхода [3]. Такой подход может быть реализован путем формирования общих фармакопейных стандартов качества на ЛС и последовательной гармонизацией фармакопейных статей (общих и частных) национальных фармакопей государств — членов ЕАЭС4.
В соответствии с Концепцией гармонизации фармакопей государств — членов ЕАЭС5 в качестве базовой (основной фармакопеи первого уровня) признана Европейская фармакопея, а статусом фармакопеи второго уровня обладают Британская фармакопея и Фармакопея США. Подготовка фармакопейных текстов для Фармакопеи Союза осуществляется исходя из данного положения. Важно отметить, что ГФ РФ по принципу формирования стандартов на БЛП значительно отличается как от Европейской фармакопеи, так и от национальных фармакопей государств — членов ЕАЭС. Формирование единых подходов к разработке соответствующих фармакопейных текстов (общих фармакопейных статей (ОФС) и фармакопейных статей (ФС)) представляет собой актуальную задачу. В статье отражены основные этапы работы, проведенной авторами, по фармакопейной стандартизации БЛП в рамках Фармакопеи Союза и ГФ РФ.
Цель работы — систематизация требований к БЛП в рамках Фармакопеи Союза и ГФ РФ для гармонизации региональных и национальных стандартов.
Фармацевтические рынки на территории государств — членов ЕАЭС имеют национальные особенности, особенно заметные в подходах к формированию фармакопейных требований к БЛП. Так, Государственная фармакопея Республики Казахстан6, изданная с разрешения Европейского директората по качеству лекарственных средств и здравоохранения Совета Европы (European Directorate for the Quality of Medicines & HealthCare, Council of Europe), включает фармакопейные тексты, основанные на текущем издании Европейской фармакопеи, а также учитывает основные принципы гармонизации с Фармакопеей США и Британской фармакопеей [4]. В нее включены 6 общих статей на группы иммунобиологических лекарственных препаратов (ИБЛП), а также 15 монографий на вакцины и ЛС из плазмы крови человека.
В Государственной фармакопее Республики Беларусь7 (ГФ РБ), которая также разработана на основе Европейской фармакопеи, широко представлены ОФС, касающиеся требований к БЛП и методам их контроля. Однако для ГФ РБ характерно также наличие дополнительных статей в соответствии с национальными подходами к стандартизации БЛП, например по определению биологической активности инсулина в испытаниях на животных8.
ГФ РФ в значительной степени сохранила преемственность требований к БЛП, методам их контроля и биологическим испытаниям, установленным ранее в Государственной фармакопее СССР. Методы контроля качества БЛП в течение десятилетий были регламентированы различными методическими указаниями, а также ФС на физико-химические, химические, физические и иммунохимические методы контроля медицинских ИБЛП9 и ФС на соответствующие лекарственные препараты. В ГФ РФ XIII изд. дополнительно были включены ОФС, касающиеся требований к группам БЛП и методов их контроля, а также ФС на конкретные БЛП [5][6]. В ГФ РФ XIV изд. этот перечень был значительно расширен.
Фармакопейные стандарты представлены 25 ОФС на группы БЛП (рис. 1), которые характеризуются многократным дублированием нормативных требований. Например, ОФС.1.7.1.0010.18 «Биологические лекарственные препараты» содержит требования к ИБЛП и биотехнологическим лекарственным препаратам, которые повторно встречаются в ОФС.1.7.1.0018.18 «Иммунобиологические лекарственные препараты» и ОФС.1.7.1.0011.18 «Биотехнологические лекарственные препараты». Такой подход в формировании стандартов качества на БЛП с одной стороны акцентирует внимание на ключевых требованиях к БЛП, с другой стороны делает фармакопейные тексты объемными и инертными в плане своевременной актуализации.
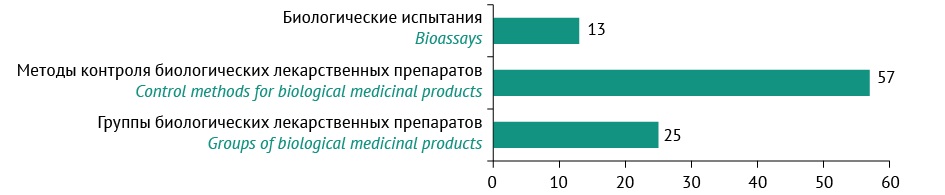
Рисунок подготовлен авторами / The figure is prepared by the authors
Рис. 1. Количество общих фармакопейных статей на биологические лекарственные препараты и методы их контроля в Государственной фармакопее Российской Федерации XIV изд.
Fig. 1. Number of general chapters for biological medicinal products and methods for their control in the State Pharmacopoeia of the Russian Federation, 14 th ed.
В рамках фармакопейной стандартизации ИБЛП, препаратов из плазмы крови человека, пробиотиков, бактериофагов и ряда других БЛП в ГФ РФ XIV изд. включено 100 ФС (рис. S1, опубликован на сайте журнала, https://doi.org/10.30895/2221-996X-2025-25-1-71-82-fig-s1). Часть из них, например на лекарственные препараты пробиотиков, бактериофагов, пирогенал, не имеют аналогов в ведущих мировых фармакопеях, тогда как другие представляют собой фармакопейные стандарты, негармонизированные с аналогичными в других фармакопеях. Эти стандарты могут отличаться как в сторону ужесточения требований (например, наличие дополнительных показателей качества), так и в сторону их ослабления (менее жесткие нормы, отсутствие необходимых показателей качества).
В мировой фармакопейной практике определение и классификация БЛП неоднозначны и представлены лишь в немногих фармакопеях. Так, Фармакопея США определяет БЛП как «любой вирус, терапевтическая сыворотка, токсин, антитоксин, или другой аналогичный продукт, который может использоваться для профилактики и лечения заболеваний или травм у людей10», производимый по лицензии в соответствии с законодательством США (Свод законов США)11. В фармакопейных целях12 термин «биологические препараты» применим к лекарственным препаратам, лицензированным в соответствии со Сводом законов США13 и удовлетворяющим требованиям Управления по контролю за качеством продуктов питания и лекарственных средств США (Food and Drug Administration, FDA)14. Свод законов США устанавливает следующий перечень БЛП15:
Согласно Китайской фармакопее17 к профилактическим биологическим продуктам относятся бактериальные и вирусные вакцины, а к терапевтическим — антитоксины и антисыворотки, продукты крови, биотехнологические продукты. В фармакопее Таиланда18 БЛП определяются как продукты биологического происхождения, включая продукты животного, бактериального или растительного происхождения (вакцины, диагностические препараты), а также животного или человеческого происхождения (антисыворотки, препараты крови), эффективность и безопасность которых невозможно оценить исключительно химическими или физическими методами испытаний.
Международная фармакопея не использует термин «биологические лекарственные препараты», однако Всемирная организация здравоохранения (ВОЗ) классифицирует БЛП как ЛС, получаемые путем культивирования и последующей очистки из клеточных культур бактерий, дрожжей или животных клеток. К этой группе относятся вакцины, факторы роста, иммуномодуляторы, моноклональные антитела, а также продукты, полученные из крови и плазмы крови человека19. Контроль качества и регулирование обращения БЛП отличаются от аналогичных процессов для других групп лекарственных препаратов из-за особенностей их природы и способов производства. Каждая серия БЛП подвергается строгому контролю на каждом этапе производства, обеспечивая соответствие предыдущим сериям. Применение стандартных образцов (СО) соответствующей квалификации (международных, фармакопейных) дополнительно гарантирует стабильность качества в рамках одного производства между сериями, а также позволяет сравнивать БЛП разных производителей и стран20. Установление универсальных требований к исходному сырью, материалам, процессам производства, а также к качеству конечного продукта играет важную роль в обеспечении эффективности и безопасности БЛП21.
Обоснование общих понятий и положений для биологических лекарственных препаратов
Разработка проекта ОФС, касающегося требований к БЛП, в рамках Фармакопеи Союза, в первую очередь основывалась на принципах гармонизации с учетом национальных особенностей ГФ РФ как единственной фармакопеи среди государств — членов ЕАЭС, содержащей обширный перечень стандартов качества на БЛП, включая ОФС.1.7.1.0010.18 «Биологические лекарственные препараты»22. В то же время национальное законодательство Российской Федерации значительно отличается от регионального (ЕАЭС) в части определения понятия «биологический лекарственный препарат». Так, согласно статье 4 Федерального закона № 61-ФЗ23 для БЛП установлены два основных признака: наличие биологического источника для производства или выделения, а также использование для контроля качества комбинации биологических и физико-химических методов. В Правилах проведения исследований биологических лекарственных средств ЕАЭС24 определение БЛП дополнено необходимостью оценки производственного процесса и методов его контроля. В фармакопейных целях в рамках ЕАЭС к БЛП отнесены лекарственные препараты, действующее (активное) вещество которых произведено или выделено из биологического источника, и для характеристики их свойств обычно применяют комплекс биологических и физико-химических методов анализа совместно с оценкой производственного процесса и методов его контроля.
С целью устранения несоответствий с национальным законодательством, а также разночтений в классификации БЛП в проекте ОФС «Биологические лекарственные препараты» нами предложен подход к группированию этих лекарственных препаратов по различным классификационным признакам:
Различные подходы к стандартизации лекарственных препаратов антибиотиков природного и полусинтетического происхождения25 обусловили решение не включать их в группу БЛП в рамках Фармакопеи Союза, несмотря на получение путем ферментации (с возможной последующей химической модификацией). Кроме того, на препараты из крови, плазмы и клеток крови человека (за исключением плазмы крови человека, приготовленной по методу, включающему промышленный процесс) не распространяются требования фармакопейных стандартов; для них действуют иные регуляторные акты в рамках национального законодательства.
Формирование общих требований к производству биологических лекарственных препаратов
Выбор объема фармацевтических, биологических, доклинических и клинических исследований для оценки БЛП должен проводиться с учетом особенностей исходного сырья и технологического процесса производства БЛП в соответствии с актами Евразийской экономической комиссии в сфере регулирования обращения биологических лекарственных средств26.
Производство БЛП характеризуется рядом особенностей, связанных с уникальными технологическими процессами (например, культивирование производственных штаммов микроорганизмов или штаммов-продуцентов эукариотических клеточных культур, экстракция веществ из биологических тканей и крови человека и животных и др.) и вариабельностью свойств исходного сырья. Это требует особого внимания к обеспечению стабильности характеристик лекарственного препарата, включая состав и природу родственных и производственных примесей27. Производственный процесс БЛП регулируется как Правилами надлежащей производственной практики28, так и особыми правилами и рекомендациями, что гарантирует надлежащее качество лекарственных препаратов. Особенности технологии, критические этапы и необходимые испытания промежуточных продуктов описаны в соответствующих ФС и ОФС, устанавливающих требования к отдельным группам БЛП.
На основании положений трехстороннего руководства Международного совета по гармонизации технических требований к лекарственным средствам для медицинского применения (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, ICH29) в Фармакопее Союза принята стратегия контроля БЛП, учитывающая основные принципы обеспечения качества. Эта стратегия базируется на оценке рисков, основанной на данных контроля исходного сырья и материалов, контроле критически важных параметров на промежуточных этапах производства с использованием соответствующих методов, а также валидации производственного процесса для подтверждения стабильности выпускаемых серий БЛП, сопоставимых с сериями, имеющими доказанную клиническую эффективность и безопасность [7]. Особое внимание уделяется оценке эффективности каждого этапа очистки, направленной на удаление и/или инактивацию посторонних агентов и примесей, источником которых могут быть клетки хозяина (ДНК), штаммы-продуценты (вирусы и вирусные частицы, белки), а также удаление примесей, связанных с самим процессом производства, таких как гетерологичные белки (компоненты питательных сред, иммуносорбенты для аффинной хроматографии др.). Процесс производства БЛП из плазмы крови человека должен включать не менее двух эффективных, отличающихся по механизму действия, стадий вирусной инактивации и/или элиминации возможных контаминирующих вирусов.
Все исходное сырье и материалы животного происхождения, представляющие опасность передачи возбудителей губчатой энцефалопатии животных, требуют оценки этого риска. БЛП, полученные с использованием клеточных линий, подвержены риску вирусной контаминации, связанной с возможным заражением клеточных субстратов или случайным попаданием вирусов на различных этапах технологического процесса. Обеспечение безопасности БЛП в отношении вирусного заражения достигается соблюдением требований к субстратам производства, проведением испытаний на наличие вирусов и оценкой удаления и/или инактивации вирусов в ходе производственного процесса. Особое внимание при получении БЛП из плазмы и клеток крови, биологических жидкостей и органов человека уделяется строгому соблюдению требований к здоровью доноров.
Для предотвращения нежелательных изменений свойств БЛП, вызванных многократными пересевами или большим числом пассажей, производство БЛП, получаемых из культур микроорганизмов или клеток, базируется на системе банков посевных культур (микроорганизмов или клеток), охарактеризованных должным образом. Для культивирования микроорганизмов и клеточных линий используют подходящие питательные и культуральные среды. В технологическом процессе необходимо использовать генетически стабильные производственные штаммы бактерий и вирусов, а также охарактеризованные и депонированные в официальных коллекциях клеточные линии животного происхождения, контролируемые по биологическим свойствам. Допустимое количество генераций (удвоений, пассажей) культур микроорганизмов или клеток для получения серии готового лекарственного препарата определяется на этапе фармацевтической разработки и подтверждается при валидации процесса производства. Генетическая стабильность производственных штаммов служит критерием ограничения числа пассажей бактерий или вирусов.
Стандартные образцы в контроле качества биологических лекарственных препаратов
Для оценки биологической активности БЛП30, выраженной в международных единицах, используют международные СО, калиброванные в международных единицах. Эти СО служат основой для оценки активности или эффективности БЛП, обеспечивая согласованность результатов измерений в испытаниях лекарственных препаратов при контроле их качества с дозировкой при клиническом применении (при назначениях пациентам)31. Международные единицы устанавливаются на основе результатов испытаний кандидатов в СО в нескольких лабораториях, расположенных в разных странах, с использованием собственных методик32. В испытаниях БЛП также могут применяться фармакопейные СО, калиброванные относительно соответствующего международного СО. За международную единицу активности принимают активность определенного количества международного СО, устанавливаемую ВОЗ. Для оценки биологической активности БЛП, выражаемой в других единицах (например, единиц связывания анатоксина), используют подходящий СО, охарактеризованный надлежащим образом.
В отдельных случаях для идентификации, количественного определения и других испытаний некоторых БЛП в качестве СО используют серию лекарственного препарата с подтвержденной стабильностью и репрезентативностью относительно серий, прошедших клинические исследования, либо репрезентативную серию фармацевтической субстанции. Этот подход применяется, например, для препаратов аллергенов и моноклональных антител.
Особенности контроля качества биологических лекарственных препаратов
Установление характеристик БЛП (готового продукта) включает определение физико-химических свойств, чистоты и наличия примесей, биологической активности, безопасности и, при необходимости, иммунохимических свойств33. Допускается проведение испытаний на соответствующих промежуточных продуктах в тех случаях, когда их результаты важны для оценки качества (например, отсутствие вирусов), но не могут быть получены непосредственно на лекарственном препарате.
В ряде случаев активность БЛП зависит от конъюгации действующего вещества с другим соединением или от связывания с адъювантом. Диссоциацию активного(ых) ингредиента(ов) от носителя, используемого в составе конъюгатов или адъювантов, испытывают в реальном времени и при реальной температуре (в том числе в условиях транспортировки). Оценка стабильности34 таких БЛП может быть затруднена, так как определение физико-химических характеристик или биологической активности в испытаниях in vitro может дать неточные результаты. В таких случаях проводят, например, испытание промежуточного продукта до конъюгации/связывания или используют другие подходы.
Для испытаний БЛП используют разнообразные методы, позволяющие оценить их физико-химические и биологические свойства.
Определение физико-химических свойств БЛП включает испытания по подтверждению состава, физических свойств, первичной структуры действующего вещества, в том числе оценке структурной гетерогенности (если применимо).
Биологическая активность — один из важнейших показателей качества БЛП, характеризующий способность препарата оказывать определенный биологический эффект35. Для оценки биологической активности могут быть проведены испытания in vivo (на животных), в которых измеряют биологический ответ организма на введение БЛП, или исследования in vitro на культурах клеток, позволяющие определить биохимический или физиологический ответ на клеточном уровне. Также могут быть использованы биохимические методы, устанавливающие, например, скорость ферментативных реакций или биологические ответы, индуцируемые иммунологическими взаимодействиями. Устанавливаемое значение показателя, выражаемое в единицах активности36, представляет собой количественную меру биологической активности БЛП и связано с его релевантными биологическими свойствами. При фармацевтической разработке БЛП должна быть подтверждена сопоставимость результатов определения активности в биологических испытаниях при контроле качества с эффективностью БЛП.
Если данные о физико-химических свойствах БЛП недостаточны для подтверждения структуры более высокого порядка, допускается проведение биологических испытаний с расширенными границами доверительного интервала в сочетании с определением содержания действующего (активного) вещества. Замена биологических исследований физико-химическими допускается, если последние позволяют получить детальную характеристику БЛП, включая информацию о структуре высокого порядка, и установлена значимая корреляция с биологической активностью. Если для количественного определения биологической активности применяются исключительно физико-химические методы (основанные на установленной корреляции), их результаты выражают в массовых единицах.
В случае БЛП на основе антител проводят оценку их иммунологических свойств. Для определения аффинности, авидности и иммунореактивности (включая перекрестную реактивность) исследуют связывание антител с очищенными антигенами или участками антигенов. Результаты иммунохимического анализа могут быть использованы для установления подлинности, однородности или чистоты, а также количественного определения БЛП.
Для некоторых групп БЛП, например вакцин и анатоксинов, проводят испытание на безопасность, включая оценку вирулентности вакцинных штаммов бактерий или вирусов, а также их токсигенности. Необходимость испытания на безопасность обусловлена возможной остаточной патогенностью производственных штаммов бактерий или вирусов, которая может проявляться в виде их вирулентных и токсигенных свойств. Возможные нарушения технологического процесса производства БЛП, например недостаточная детоксикация бактериальных токсинов при изготовлении анатоксинов, могут приводить к реверсии токсических свойств используемых штаммов.
Кроме того, для БЛП применимы испытания, обязательные для определенной лекарственной формы, такие как стерильность, микробиологическая чистота и др.
Основные подходы к замене или исключению испытаний in vivo в контроле качества биологических лекарственных препаратов
Одной из проблем при оценке качества БЛП является широкое применение методов in vivo [8]. В последнее время ведутся активные исследования, направленные на замену методов in vivo на более этичные и эффективные методы in vitro. Это позволяет ведущим мировым фармакопеям рассматривать альтернативные методы испытаний как для количественного определения, так и для оценки безопасности БЛП. В Европейской фармакопее предусмотрена возможность количественного определения вакцины (анатоксина) для профилактики дифтерии как методом летального заражения или методом кожных проб, так и по оценке уровня антител у иммунизированных животных37. Применимость методов на разных этапах жизненного цикла данной вакцины четко регламентирована: методы, основанные на определении защитного действия анатоксина при заражении дифтерийным токсином иммунизированных морских свинок, используются на стадии разработки анатоксина и при валидации (ревалидации) производственного процесса; метод, основанный на определении титра противодифтерийных антитоксических антител в сыворотке крови иммунизированных морских свинок, — для контроля качества выпускаемого анатоксина. Аналогичным образом рассматриваются стратегии контроля качества вакцины (анатоксина) для профилактики столбняка38, вакцины для профилактики гепатита В (рекомбинантной)39 и гепатита А40. Из-за большой вариабельности методов in vivo их возможная замена на методы in vitro является сложной задачей. В связи с этим особое значение приобретает оценка сопоставимости результатов испытаний, полученных с применением более стабильных методов in vitro.
В настоящее время при участии некоторых фармакопейных комиссий проводится активная работа по научно обоснованному исключению испытания на аномальную токсичность из спецификаций на БЛП. Ряд мировых фармакопей полностью исключили это испытание. Так, Европейская фармакопея, начиная с отказа от посерийного контроля субстанций и лекарственных препаратов на аномальную токсичность, в настоящее время полностью исключила такие испытания. В рамках Фармакопеи США вместо отдельной монографии по испытаниям на аномальную токсичность ранее использовалась методика определения общей безопасности, указанная в Своде законов США41, которая также в настоящее время исключена.
Эксперты Индийской фармакопейной комиссии приняли за основу стратегию, предложенную ВОЗ, предполагающую возможность исключения таких испытаний при условии, что производитель установит стабильность процесса, однородность характеристик препарата от серии к серии и охарактеризует каждую серию с помощью достаточного количества физико-химических и/или биологических методов, одобренных национальным регуляторным органом. Этот подход позволил исключить испытания на аномальную токсичность из 39 монографий на вакцины. В тех случаях, когда исключение данного испытания недопустимо, оно проводится на готовом нерасфасованном продукте на этапе производства.
В Японской и Корейской фармакопеях общая монография по аномальной токсичности отсутствует. Однако в частных монографиях на некоторые ЛС регламентированы требования к аномальной токсичности и присутствуют методики ее определения. Такой дифференцированный подход позволяет оценивать риск появления нерегламентированных токсичных примесей для конкретного ЛС с учетом характеристик исходного сырья и особенностей технологии производства. Так, например, в обеих указанных фармакопеях для урокиназы и хорионического гонадотропина, получаемых из сырья человеческого происхождения, предусмотрены испытания на аномальную токсичность.
С учетом современной тенденции обоснованного исключения испытаний на аномальную токсичность в проект ОФС «Биологические лекарственные препараты» для Фармакопеи Союза такие испытания не включены в качестве обязательной процедуры стандартизации БЛП.
Следует отметить, что в настоящее время в мировом фармакопейном сообществе активно обсуждается вопрос полного исключения испытания на пирогенность с использованием кроликов42. Согласно новыми требованиями Европейской фармакопеи в испытании на пирогенные вещества лекарственных препаратов, в том числе БЛП, необходимо учитывать возможную контаминацию бактериальными эндотоксинами или пирогенными веществами неэндотоксиновой природы. В зависимости от этого осуществляется выбор метода испытаний на бактериальные эндотоксины или тест активации моноцитов. Испытания на пирогенность с использованием кроликов при этом исключены как фармакопейный метод43.
При подготовке проекта ОФС «Биологические лекарственные препараты» для Фармакопеи Союза с учетом отсутствия у производителей из стран ЕАЭС опыта полной замены испытаний на кроликах методом активации моноцитов было принято решение рассматривать данные испытания в рамках ОФС на отдельные группы БЛП.
В соответствии с установленными современными принципами стандартизации БЛП на региональном уровне на основе подготовленного нами и утвержденного Фармакопейным комитетом ЕАЭС проекта соответствующей ОФС сформирована обновленная структура раздела по БЛП для ГФ РФ XV изд., а также актуализирована ОФС.1.7.1.0010.18 «Биологические лекарственные препараты». В отличие от ОФС.1.7.1.0010.18 в ГФ РФ XIV изд. в подготовленном проекте БЛП сгруппированы по нескольким классификационным признакам, что позволяет в полной мере охватить все БЛП. Из рассматриваемой ОФС исключены указания о порядке регистрации фармацевтических субстанций для производства БЛП, а также о соответствии производства требованиям надлежащей производственной практики.
Требования для конкретных БЛП формируются с учетом информации, представленной как в ФС, так и в ОФС, регламентирующих требования к отдельным группам БЛП, и ОФС «Биологические лекарственные препараты». Это позволило исключить дублирование информации. Учитывая многообразие группы БЛП, исключено перечисление показателей качества как для фармацевтических субстанций, так и для лекарственных препаратов. При этом в ОФС включены наиболее важные параметры качества (например, активность) и принципы их нормирования и контроля. К включению в ГФ РФ XV изд. запланированы 13 ОФС, касающихся требований к различным группам БЛП (рис. S2, опубликован на сайте журнала, https://doi.org/10.30895/2221-996X-2025-25-1-71-82-fig-s2).
Проекты ОФС «Вакцины и анатоксины», «Иммуноглобулины человека», «Лекарственные препараты аллергенов для медицинского применения», «Иммуноглобулины и иммунные сыворотки гетерологичные», «Лекарственные средства, получаемые с использованием технологии рекомбинантной ДНК», «Моноклональные антитела для медицинского применения» гармонизированы с соответствующими фармакопейными текстами, рассмотренными и утвержденными на Фармакопейном комитете ЕАЭС44. ОФС «Токсины ботулинические», «Эритропоэтины», «Филграстимы» будут представлены только в ГФ РФ, однако при их подготовке учтены региональные требования в рамках стандартизации БЛП.
Исключение дублирования требований к БЛП в ОФС, касающихся различных групп БЛП, а также особенности формирования стандартов качества в рамках фармакопейных статей требуют нового подхода к применению всей совокупности информации в фармакопейных стандартах качества. Этот подход учитывает основные требования к соответствующим лекарственным формам, вспомогательным веществам, упаковке, материалам, используемым при производстве упаковки, и др. Так, для оценки качества препарата иммуноглобулина человека необходимо учитывать требования ФС на конкретный специфический иммуноглобулин (при необходимости), ФС на иммуноглобулин человека в зависимости от способа введения (внутривенное, подкожное или внутримышечное), ОФС «Иммуноглобулины человека», а также ОФС «Биологические лекарственные препараты». Требования ОФС «Иммуноглобулины человека», включая испытания лекарственного препарата, должны быть дополнены требованиями, представленными в ФС. Аналогичный подход применяется при формировании требований к иммуноглобулинам и иммунным сывороткам гетерологичным. В то же время для вакцин и анатоксинов, в силу больших различий внутри этих групп препаратов, требования ФС включают (а не дополняют), испытания, указанные в ОФС «Вакцины и анатоксины».
Проведенный анализ международных и национальных нормативных и правовых документов позволил сформулировать определение БЛП, применяемое в фармакопейных целях, как для Фармакопеи Союза, так и для Государственной фармакопеи Российской Федерации. Проведена фармакопейная стандартизации требований к различным группам БЛП, выделены основные аспекты, требующие рассмотрения в рамках производства (технологии), контроля качества промежуточных продуктов и лекарственного препарата.
В рамках гармонизации Государственной фармакопеи Российской Федерации с требованиями Фармакопеи Союза установлены основные требования к БЛП с учетом особенностей их производства и контроля качества.
Разработан проект общей фармакопейной статьи «Биологические лекарственные препараты» для Фармакопеи Союза, и на его основе актуализирована ОФС.1.7.1.0010.18 «Биологические лекарственные препараты». Обоснованы подходы к формированию фармакопейных статей и общих фармакопейных статей, касающихся требований к различным группам БЛП.
Дополнительная информация. На сайте журнала «БИОпрепараты. Профилактика, диагностика, лечение» размещены рисунки S1 и S2.
https://doi.org/10.30895/2221-996X-2025-25-1-71-82-fig-s1
https://doi.org/10.30895/2221-996X-2025-25-1-71-82-fig-s2
Вклад авторов. Все авторы подтверждают соответствие своего авторства критериям ICMJE. Наибольший вклад распределен следующим образом: О.Г. Корнилова — концепция работы, написание текста рукописи, формулировка выводов; В.Л. Багирова — концепция работы, формулировка выводов, утверждение окончательной версии статьи для публикации.
Additional information. Figures S1 and S2 are published on the website of Biological Products. Prevention, Diagnosis, Treatment.
https://doi.org/10.30895/2221-996X-2025-25-1-71-82-fig-s1
https://doi.org/10.30895/2221-996X-2025-25-1-71-82-fig-s2
Authors’ contributions. All the authors confirm that they meet the ICMJE criteria for authorship. The most significant contributions were as follows. O.G. Kornilova conceptualised the study, drafted the manuscript, and formulated the conclusions. V.L. Bagirova conceptualised the study, formulated the conclusions, and approved the final version of the manuscript for publication.
1. Global Biologics Market Report. Value Market Research; 2024.
2. Global Biopharmaceuticals Market Size, Share, and Trends Analysis Report — Industry Overview and Forecast to 2031. Data Bridge Market Research; 2025.
3. Федеральный закон от 25.10.2016 № 5-ФЗ «О ратификации соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза».
4. Решение Коллегии Евразийской экономической комиссии от 22.09.2015 № 119 «О Концепции гармонизации фармакопей государств — членов Евразийского экономического союза».
5. Там же.
6. Государственная фармакопея Республики Казахстан. Т. 1. Алматы: Издательский дом «Жибек жолы»; 2008.
Государственная фармакопея Республики Казахстан. Т. 2. Алматы: Издательский дом «Жибек жолы»; 2009.
7. Государственная фармакопея Республики Беларусь: в 2 т. Т. 1. Общие методы контроля лекарственных средств. Министерство здравоохранения Республики Беларусь, УП «Центр экспертиз и испытаний в здравоохранении»; Шерякова АА, ред. Молодечно: Типография Победа; 2012.
Государственная фармакопея Республики Беларусь: в 2 т. Т. 2. Контроль качества субстанций для фармацевтического использования и лекарственного растительного сырья. Министерство здравоохранения Республики Беларусь, УП «Центр экспертиз и испытаний в здравоохранении». Марченко СИ, ред. Молодечно: Типография Победа; 2016.
8. ОФС.2.7.50 Определение биологической активности инсулина. Государственная фармакопея Республики Беларусь. Т. 2; 2016.
9. ФС.42-344ВС-90 Физико-химические, химические, физические и иммунохимические методы контроля медицинских иммунобиологических препаратов. М.: Типография Министерства здравоохранения СССР; 1990.
ФС.42-3874-99 Физико-химические, химические, физические и иммунохимические методы контроля медицинских иммунобиологических препаратов. М.: Типография Министерства здравоохранения РФ; 2000.
10. <1041> Biologics. United State Pharmacopeia; 2024.
11. Public Health Service Act, 42 US Code, Chapter 6A; 2015.
12. <1041> Biologics. United State Pharmacopeia; 2024.
13. Public Health Service Act, 42 US Code, Chapter 6A; 2015.
14. Code of Federal Regulations, Title 21, Parts 600–680.
15. Public Health Service Act, 42 US Code, Chapter 6A; 2015.
16. Code of Federal Regulations, Title 21, Chapter I, Subchapter F, Part 600, Subpart A, § 600.3.
17. General Notices. Pharmacopoeia of the Peoples’ Republic of China. Vol. 3; 2020.
18. https://www.bdn.go.th/tp/ebook/qQycAKtmpR9gC3q0GT5gMJq0qT5co3uw
19. WHO Good manufacturing practices for biological products. Proposed replacement of Technical report series No. 822, Annex 1. WHO; 2015.
https://www.who.int/health-topics/biologicals#tab=tab_1
20. https://www.who.int/health-topics/biologicals#tab=tab_1
21. Там же.
22. ОФС.1.7.1.0010.18 Биологические лекарственные препараты. Государственная фармакопея Российской Федерации. XIV изд. Т. 2; 2018.
23. Федеральный закон от 12.04.2010 № 61-ФЗ «Об обращении лекарственных средств».
24. Решение Совета Евразийской экономической комиссии от 03.11.2016 № 89 «Об утверждении Правил проведения исследований биологических лекарственных средств Евразийского экономического союза».
25. https://www.who.int/health-topics/biologicals#tab=tab_1
26. Решение Совета Евразийской экономической комиссии от 03.11.2016 № 89 «Об утверждении Правил проведения исследований биологических лекарственных средств Евразийского экономического союза».
27. ICH Q5A(R2) Viral safety evaluation of biotechnology products derived from cell lines of human or animal origin. ICH; 2023.
ICH Q5B Analysis of the expression construct in cells used for production of r-DNA derived protein products. ICH; 1995.
ICH Q5D Derivation and characterisation of cell substrates used for production of biotechnological/biological product. ICH; 1997.
ICH Q6B Specifications: Test procedures and acceptance criteria for biotechnological/biological products. ICH; 1999.
28. Решение Совета Евразийской экономической комиссии от 03.11.2016 № 77 «Об утверждении Правил надлежащей производственной практики Евразийского экономического союза».
29. ICH Q6B Specifications: Test procedures and acceptance criteria for biotechnological/biological products. ICH; 1999.
30. https://www.who.int/health-topics/biologicals#tab=tab_1
31. https://www.who.int/activities/providing-international-biological-reference-preparations
32. WHO Expert Committee on Biological Standardization. 55th report. Technical report series No. 932. WHO; 2004.
33. ICH Q6B Specifications: Test procedures and acceptance criteria for biotechnological/biological products. ICH; 1999.
34. ICH Q5C Quality of biotechnological products: Stability testing of biotechnological/biological Products. ICH; 1995.
35. ICH Q6B Specifications: Test procedures and acceptance criteria for biotechnological/biological products. ICH; 1999.
36. Там же.
37. 2.7.6. Assay of diphtheria vaccine (adsorbed). European Pharmacopoeia. 11th ed. (Suppl. 11.7).
38. 2.7.8. Assay of tetanus vaccine (adsorbed). European Pharmacopoeia. 11th ed. (Suppl. 11.7).
39. 2.7.15. Assay of hepatitis B vaccine (rDNA. European Pharmacopoeia. 11th ed. (Suppl. 11.7).
40. 2.7.14. Assay of hepatitis A vaccine. European Pharmacopoeia. 11th ed. (Suppl. 11.7).
41. Code of Federal Regulations, Title 21, Chapter I, Subchapter F, Part 610, Subpart A, § 610.11. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-F/part-610
42. Guidelines on the phasing out of animal tests for the quality control of biological products (draft version). WHO; 2024.
43. Pharmeuropa. 2023; 35.1.
44. https://eec.eaeunion.org/comission/department/deptexreg/formirovanie-obshchikh-rynkov/pharmacopoeia/farmakopeya-16-08-24.php
https://eec.eaeunion.org/comission/department/deptexreg/formirovanie-obshchikh-rynkov/pharmacopoeia/farmakopeya-26-06.php
https://eec.eaeunion.org/comission/department/deptexreg/formirovanie-obshchikh-rynkov/pharmacopoeia/farmakopeya-15-06.php
https://eec.eaeunion.org/comission/department/deptexreg/formirovanie-obshchikh-rynkov/pharmacopoeia/farmakopeya-14-02.php
1. Меркулов ВА, Ягудина РИ, Серпик ВГ. Глобальный спектр разработки инновационных лекарственных препаратов: описательный обзор. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2024;14(1):14–28. https://doi.org/10.30895/1991-2919-2024-14-1-14-28
2. Устюгова ЕА, Савкина МВ, Горяев АА, Бондарев ВП, Меркулов ВА, Мельникова ЕВ. Применение биомедицинских клеточных продуктов для лечения онкологических заболеваний. БИОпрепараты. Профилактика, диагностика, лечение. 2019;19(4):206–14. https://doi.org/10.30895/2221-996X-2019-19-4-206-214
3. Тулегенова АУ. Фармакопея Евразийского экономического союза: испытания, методики и требования. Вестник Росздравнадзора. 2021;(4):64–8. EDN: NIABUH
4. Мусинов СР, Тулегенова АУ. Государственная фармакопея — главный стандарт качества лекарственных средств и изделий медицинского назначения в Республике Казахстан. Ведомости Научного центра экспертизы средств медицинского применения. 2016;(2):26–30. EDN: WBVVUT
5. Цындымеев АГ, Олефир ЮВ, Меркулов ВА, Саканян ЕИ. Российская фармакопейная практика и перспективы ее развития. Ведомости Научного центра экспертизы средств медицинского применения. 2016;(2):4–7. EDN: WBVVTF
6. Мовсесянц АА, Бондарев ВП, Олефир ЮВ, Меркулов ВА, Шимчук ЛФ. Стандарты качества иммунобиологических лекарственных препаратов — новое в Государственной фармакопее Российской Федерации. Ведомости Научного центра экспертизы средств медицинского применения. 2016;(2):38–41. EDN: WBVVVX
7. Солдатов АА, Яковлев АК, Авдеева ЖИ, Горенков ДВ, Коровкин АС, Косенко ВВ. Обзор современных регуляторных требований к изучению стабильности биологических лекарственных препаратов. БИОпрепараты. Профилактика, диагностика, лечение. 2024;24(3):335–47. https://doi.org/10.30895/2221-996X-2024-24-3-335-347
8. Viviani L, Halder M, Gruber M, Bruckner L, Cussler K, Sanyal G, et al. Global harmonization of vaccine testing requirements: Making elimination of the ATT and TABST a concrete global achievement. Biologicals. 2020;63:101–5. https://doi.org/10.1016/j.biologicals.2019.10.007
Корнилова Ольга Геннадьевна, д-р фарм. наук
Петровский б-р, д. 8, стр. 2, Москва, 127051
Багирова Валерия Леонидовна, д-р фарм. наук, проф.
Петровский б-р, д. 8, стр. 2, Москва, 127051

|
1. Рис. S1. Количество фармакопейных статей на биологические лекарственные препараты в Государственной фармакопее Российской Федерации XIV изд. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(252KB)
|
Метаданные ▾ | |
|
|
2. Рис. S2. Общие фармакопейные статьи (ОФС) на группы биологических лекарственных препаратов, планируемые к включению в Государственную фармакопею Российской Федерации XV изд. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(428KB)
|
Метаданные ▾ | |
Корнилова О.Г., Багирова В.Л. Фармакопейная стандартизация биологических лекарственных препаратов: основные принципы в условиях единого фармацевтического рынка стран Евразийского экономического союза. БИОпрепараты. Профилактика, диагностика, лечение. 2025;25(1):71-82. https://doi.org/10.30895/2221-996X-2025-25-1-71-82
Kornilova O.G., Bagirova V.L. Pharmacopoeial standardisation of biological medicinal products: Basic principles for the common pharmaceutical market of the Eurasian Economic Union. Biological Products. Prevention, Diagnosis, Treatment. 2025;25(1):71-82. (In Russ.) https://doi.org/10.30895/2221-996X-2025-25-1-71-82

Издатель: ФГБУ «НЦЭСМП» Минздрава России































